

1 引言
数学模型为结核病的定性和定量研究提供了可靠的理论工具. 目前, 基于肺结核实际病例评估结核病的传播风险及预测其流行趋势已相当广泛[3⇓-5]. 但由于数据收集和技术处理等限制, 模型预测的可靠性和准确性有待提高. 参数辨识性是依据输出变量判别模型参数是否唯一的主要研究方法. 如果模型参数集可以从给定的系统输入和可测量的系统输出中唯一确定, 则认为模型是可辨识的, 否则称为不可辨识. 目前, 关于常微分系统模型的参数辨识性研究已有许多方法, 如微分代数法, 幂级数展开法, 相似变换法等[6]. 但由于传染病受到社交网络、年龄结构、病程及环境等因素的影响, 建立具有结构性的偏微分系统更具现实意义. 由于肺结核病具有较长的潜伏期, Cao 和 Gao 建立如下具有病程结构和复发的肺结核病模型 (1.1)[7](参见流程图 1)
其中,
图 1

由于肺结核病潜伏期到感染期进程缓慢, 因此将潜伏类到感染类的转化速率定义为一类依赖于年龄的函数
其中,

模型 (1.1) 的初始条件为
定义1.1 模型 (1.1) 的基本再生数为
其中,
根据文献 [5,定理 1-3] 有如下引理
引理1.1 当
若模型的参数是可辨识的, 则意味着由模型输出可以唯一确定一组参数值. 模型 (1.1) 是由常微分和偏微分方程组成的混合系统, 且模型中部分参数依赖病程
2 模型参数的结构辨识性分析
模型参数的结构辨识性是指通过输入输出数据 (不考虑数据噪声) 确定模型的参数是否唯一的方法, 因此称为先验可辨识性分析. 假设模型满足如下系统
其中
定义2.1[6] 对于系统 (2.1) 的任意两个参数集
其中,
这里
其中
其中,
如果
(i) 计算敏感性矩阵乘积
(ii) 计算矩阵
(iii) 将特征值按照从小到大排序;
(iv) 寻找步骤 (iii) 中绝对值最小的特征值及其特征向量中绝对值最大分量, 定义该分量对应的参数为最不可辨识的参数;
(v) 重复步骤 (iv) 将其他参数进行排序, 即可得到参数辨识可能性的排序.
由于报道病例和实际感染病例间存在误差, 我们引入参数
事实上, 矩阵 (2.4) 的对角线元素表征参数
观察发现, 参数辨识可能性的排序依次为
3 数据拟合和参数选择
我们选取公共卫生数据中心提供的中国 2010--2018 年肺结核病例数为研究对象. 依据 2011 年中国统计年鉴[10]计算得总人口为
结合 Matlab 程序拟合数据, 其中
图 2


4 实用辨识性分析
其中, 随机变量
图 3

为了定量评价不同误差水平对参数估计的影响, 我们通过求解最优化问题(3.1) 生成参数集
这里取

通常, 参数的 ARE 水平用于评估模型参数的实用辨识性. 表 3 表明参数
其中,
矩阵 (4.4) 中对角线元素依次表征参数
5 敏感性分析
敏感性分析研究数学模型或系统输入的不确定性对输出结果的影响, 是衡量模型参数变化对传染病动力学行为影响的有用工具, 尤其是对于量化疾病控制策略的预期疗效至关重要[15]. 该方法已被广泛用于常微分组成的传染病动力学模型, 本节对具有初边值条件的年龄结构传染病模型进行敏感性分析.
5.1 局部敏感性分析
引理 1.1表明系统 (1.1) 展现出阈值动力学, 其动力学性态完全由
本小节主要考虑
若

从表 4 可以看出, 基本再生数和传染率
5.2 全局敏感性分析
从图 4 可以发现, 不论对于染病群体
图 4
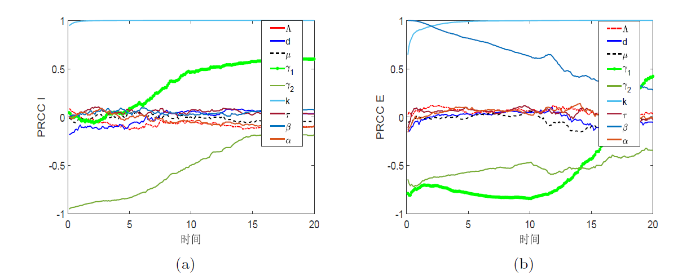
此外, 在肺结核病感染初期提高潜伏个体的恢复率
此外, 从图 4(b) 注意到, 随着肺结核病的发展, 传染率
6 总结
模型的参数辨识是通过观测模型结构并依据数据分析, 估计模型中的未知参数的数值. 在实际中, 由于模型的复杂性及数据数量和噪声等因素, 通常参数是不可辨识的, 那么预测结果可能不准确. 尽管目前已有许多针对常微分系统参数辨识的方法, 但鲜有针对年龄结构偏微分系统参数辨识的结论[8].
本文首先利用特征值方法对模型中参数进行结构辨识性分析, 依据辨识结果对不可辨识参数的可能性排序. 其次利用中国肺结核病数据拟合一类具有复发和年龄结构的肺结核病模型. 接着, 通过蒙特卡洛模拟及计算参数的相对误差值 (ARE), 得出在控制其他参数后, 我们研究的参数都是实用可辨识的. 这也验证了拟合结果的正确性. 最后, 通过局部敏感性分析和全局敏感性分析 (PRCC) 得知, 传染率
本文虽然利用特征值方法, Fisher 熵理论及蒙特卡洛模拟评估了模型的可辨识性, 但没有考虑有限元差分结构对模型误差收敛阶数及收敛速度. 另外, 尽管模型结构的复杂性为参数的结构辨识性带来挑战, 但主成分分析法可以规避一些结构辨识性计算问题[9]. 因此, 我们接下来继续讨论模型的差分格式的精度和参数结构可辨识性等方面的问题.
参考文献
Modeling relapse in infectious diseases
An integro-differential equation is proposed to model a general relapse phenomenon in infectious diseases including herpes. The basic reproduction number R(0) for the model is identified and the threshold property of R(0) established. For the case of a constant relapse period (giving a delay differential equation), this is achieved by conducting a linear stability analysis of the model, and employing the Lyapunov-Razumikhin technique and monotone dynamical systems theory for global results. Numerical simulations, with parameters relevant for herpes, are presented to complement the theoretical results, and no evidence of sustained oscillatory solutions is found.
Mathematical models of tuberculosis reactivation and relapse
DOI:10.3389/fmicb.2016.00669
PMID:27242697
[本文引用: 1]

The natural history of human infection with Mycobacterium tuberculosis (Mtb) is highly variable, as is the response to treatment of active tuberculosis. There is presently no direct means to identify individuals in whom Mtb infection has been eradicated, whether by a bactericidal immune response or sterilizing antimicrobial chemotherapy. Mathematical models can assist in such circumstances by measuring or predicting events that cannot be directly observed. The 3 models discussed in this review illustrate instances in which mathematical models were used to identify individuals with innate resistance to Mtb infection, determine the etiologic mechanism of tuberculosis in patients treated with tumor necrosis factor blockers, and predict the risk of relapse in persons undergoing tuberculosis treatment. These examples illustrate the power of various types of mathematic models to increase knowledge and thereby inform interventions in the present global tuberculosis epidemic.
On the global stability of an age-structured tuberculosis transmission model with relapse
On identifiability of nonlinear ODE models and applications in viral dynamics
Ordinary differential equations (ODE) are a powerful tool for modeling dynamic processes with wide applications in a variety of scientific fields. Over the last 2 decades, ODEs have also emerged as a prevailing tool in various biomedical research fields, especially in infectious disease modeling. In practice, it is important and necessary to determine unknown parameters in ODE models based on experimental data. Identifiability analysis is the first step in determing unknown parameters in ODE models and such analysis techniques for nonlinear ODE models are still under development. In this article, we review identifiability analysis methodologies for nonlinear ODE models developed in the past one to two decades, including structural identifiability analysis, practical identifiability analysis and sensitivity-based identifiability analysis. Some advanced topics and ongoing research are also briefly reviewed. Finally, some examples from modeling viral dynamics of HIV, influenza and hepatitis viruses are given to illustrate how to apply these identifiability analysis methods in practice.
The dynamics of an age-structured TB transmission model with relapse
Structural and practical identifiability issues of immuno-epidemiological vector-host models with application to rift valley fever
In this article, we discuss the structural and practical identifiability of a nested immuno-epidemiological model of arbovirus diseases, where host-vector transmission rate, host recovery, and disease-induced death rates are governed by the within-host immune system. We incorporate the newest ideas and the most up-to-date features of numerical methods to fit multi-scale models to multi-scale data. For an immunological model, we use Rift Valley Fever Virus (RVFV) time-series data obtained from livestock under laboratory experiments, and for an epidemiological model we incorporate a human compartment to the nested model and use the number of human RVFV cases reported by the CDC during the 2006-2007 Kenya outbreak. We show that the immunological model is not structurally identifiable for the measurements of time-series viremia concentrations in the host. Thus, we study the non-dimensionalized and scaled versions of the immunological model and prove that both are structurally globally identifiable. After fixing estimated parameter values for the immunological model derived from the scaled model, we develop a numerical method to fit observable RVFV epidemiological data to the nested model for the remaining parameter values of the multi-scale system. For the given (CDC) data set, Monte Carlo simulations indicate that only three parameters of the epidemiological model are practically identifiable when the immune model parameters are fixed. Alternatively, we fit the multi-scale data to the multi-scale model simultaneously. Monte Carlo simulations for the simultaneous fitting suggest that the parameters of the immunological model and the parameters of the immuno-epidemiological model are practically identifiable. We suggest that analytic approaches for studying the structural identifiability of nested models are a necessity, so that identifiable parameter combinations can be derived to reparameterize the nested model to obtain an identifiable one. This is a crucial step in developing multi-scale models which explain multi-scale data.
Fitting parameters and therapies of ODE tumor models with senescence and immune system
DOI:10.1007/s00285-023-02000-9
PMID:37805974
[本文引用: 2]
This work is devoted to introduce and study two quasispecies nonlinear ODE systems that model the behavior of tumor cell populations organized in different states. In the first model, replicative, senescent, extended lifespan, immortal and tumor cells are considered, while the second also includes immune cells. We fit the parameters regulating the transmission between states in order to approximate the outcomes of the models to a real progressive tumor invasion. After that, we study the identifiability of the fitted parameters, by using two sensitivity analysis methods. Then, we show that an adequate reduced fitting process (only accounting to the most identifiable parameters) gives similar results but saving computational cost. Three different therapies are introduced in the models to shrink (progressively in time) the tumor, while the replicative and senescent cells invade. Each therapy is identified to a dimensionless parameter. Then, we make a fitting process of the therapies' parameters, in various scenarios depending on the initial tumor according to the time when the therapies started. We conclude that, although the optimal combination of therapies depends on the size of initial tumor, the most efficient therapy is the reinforcement of the immune system. Finally, an identifiability analysis allows us to detect a limitation in the therapy outcomes. In fact, perturbing the optimal combination of therapies under an appropriate therapeutic vector produces virtually the same results.© 2023. The Author(s).
2000年全国结核病流行病学抽样调查报告
Report on nationwide random survey for the epidemiology of tuberculosis in 2000
Parameter identifiability and optimal control of an SARS-CoV-2 model early in the pandemic
DOI:10.1080/17513758.2022.2078899
PMID:35635313
[本文引用: 1]
We fit an SARS-CoV-2 model to US data of COVID-19 cases and deaths. We conclude that the model is not structurally identifiable. We make the model identifiable by prefixing some of the parameters from external information. Practical identifiability of the model through Monte Carlo simulations reveals that two of the parameters may not be practically identifiable. With thus identified parameters, we set up an optimal control problem with social distancing and isolation as control variables. We investigate two scenarios: the controls are applied for the entire duration and the controls are applied only for the period of time. Our results show that if the controls are applied early in the epidemic, the reduction in the infected classes is at least an order of magnitude higher compared to when controls are applied with 2-week delay. Further, removing the controls before the pandemic ends leads to rebound of the infected classes.
Structural identifiability analysis of age-structured PDE epidemic models
Sensitivity analysis in an immuno-epidemiological vector-host model
DOI:10.1007/s11538-021-00979-0
PMID:34982249
[本文引用: 2]
Sensitivity Analysis (SA) is a useful tool to measure the impact of changes in model parameters on the infection dynamics, particularly to quantify the expected efficacy of disease control strategies. SA has only been applied to epidemic models at the population level, ignoring the effect of within-host virus-with-immune-system interactions on the disease spread. Connecting the scales from individual to population can help inform drug and vaccine development. Thus the value of understanding the impact of immunological parameters on epidemiological quantities. Here we consider an age-since-infection structured vector-host model, in which epidemiological parameters are formulated as functions of within-host virus and antibody densities, governed by an ODE system. We then use SA for these immuno-epidemiological models to investigate the impact of immunological parameters on population-level disease dynamics such as basic reproduction number, final size of the epidemic or the infectiousness at different phases of an outbreak. As a case study, we consider Rift Valley Fever Disease utilizing parameter estimations from prior studies. SA indicates that [Formula: see text] increase in within-host pathogen growth rate can lead up to [Formula: see text] increase in [Formula: see text] up to [Formula: see text] increase in steady-state infected host abundance, and up to [Formula: see text] increase in infectiousness of hosts when the reproduction number [Formula: see text] is larger than one. These significant increases in population-scale disease quantities suggest that control strategies that reduce the within-host pathogen growth can be important in reducing disease prevalence.© 2021. The Author(s), under exclusive licence to Society for Mathematical Biology.
一类具有 CTL 免疫反应和免疫损害的 HIV 感染动力学模型的稳定性分析
Stability analysis of an HIV infection dynamic model with CTL immune response and immune impairment
个体化治疗耐多药肺结核失败的影响因素分析
Analysis of the influencing factors of individualized treatment failure for multidrug-resistant pulmonary tuberculosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}