


引言
活性氧是细胞内线粒体呼吸链的一种重要的活性物质,可调控很多细胞功能,比如细胞分化、增殖和凋亡等[1].低浓度的活性氧能起传递氧化还原信号的作用;但当其含量超过细胞所能维持的平衡时,活性氧会引发蛋白氧化修饰,氧化的氨基酸位点包括半胱氨酸(Cys)和甲硫氨酸(Met)等[2].其中,Met氧化是一种非常重要的蛋白质翻译后修饰方式,而且是氧化环境中(如线粒体内)一类常见的蛋白修饰方式[3]. 虽然Met是蛋白质序列中占比最少的氨基酸之一,平均含量只占到1.5~2.0%[4],但它广泛分布于各种蛋白中.据相关统计显示,99%的蛋白质含有Met[5].蛋白质表面的Met氧化之后会增强蛋白的疏水性,影响蛋白正确折叠,改变蛋白质结构、功能以及稳定性[6,7].蛋白质氧化修饰与生物体的衰老和疾病也密切相关[8].
细胞色素c是位于线粒体内的具有氧化还原性质的多功能金属蛋白,它主要定位于线粒体内外膜之间,构象多变,功能众多[9].在线粒体中,细胞色素c主要发挥电子传递功能[10].但被释放到胞质时,可能引发细胞凋亡. 细胞色素c在细胞呼吸和凋亡中发挥着双重作用,其功能转换的过程受到多种生物体内蛋白质翻译后修饰的精准调控[11],其中氧化修饰对其影响较大. 因为细胞色素c蛋白构象的变化与其中心含铁卟啉环的配位变化有关. 而细胞色素c卟啉环中心的Fe原子通常在轴向分别与Met80、His18形成配位,但其结构并不稳定. 有研究发现,细胞色素c中71~85的loop区稳定性极差,在与其他物质相互作用或者所处溶液环境变化时易于打开[12].生物体内存在的活性氧能够氧化与卟啉环中铁原子配位的Met80[13,14],从而导致细胞色素c中的Met80与Fe之间配位键的断裂[15].细胞色素c中Met80处配位键的断裂与蛋白质的功能转换密切相关,细胞色素c配位发生变化之后,会由最初的在呼吸链上传递电子转变为发挥过氧化物酶作用,并由线粒体释放进入胞质中,激发内源性细胞凋亡[9,16,17].因此,明确活性氧对细胞色素c蛋白结构的影响机制对于确定该蛋白功能机制至关重要.
为了明确在活性氧作用下,细胞色素c蛋白中Met80是否优先被氧化,及其是否影响蛋白的构象转变,我们利用Met甲基选择性标记技术,结合核磁共振(NMR)方法以及圆二色谱对不同过氧化氢(H2O2)浓度下酵母细胞色素c进行研究,对该蛋白在活性氧作用下的氧化过程进行了检测分析.发现在过氧化氢作用初期,蛋白的亚砜化修饰主要发生在Met80位点,酵母细胞色素c存在一种被部分氧化修饰但又保持整体结构稳定的状态. 暗示着活性氧诱导的细胞色素c氧化可能历经多个阶段,这对进一步理解细胞色素c在氧化应激条件下的调控功能具有重要意义.
1 实验部分
1.1 实验试剂
酵母粉(Yeast extract)、蛋白胨(Tryptone)购自Oxoid公司、磷酸二氢钾(KH2PO4)、磷酸氢二钾(K2HPO4)、磷酸氢二钠(Na2HPO4)、磷酸二氢钠(NaH2PO4)、硫酸钠(Na2SO4)、氯化钠(NaCl)、葡萄糖(C6H12O6)、硫酸铵((NH4)2SO4)、铁氰化钾(K3[Fe(CN)6])、维生素B1(Vitamin B1)、H2O2购自国药集团化学试剂有限公司,甲基13C标记的Met、抗坏血酸钠(SLA)、重水(D2O)购自Sigma-Aldrich公司;异丙基-β-D-硫代半乳糖苷(IPTG)、溶菌酶(Lysozyme egg white)购自Biosharp公司;脱氧核糖核酸酶I(DNase I)购自Thermo Scientific科技有限公司;三羟甲基氨基甲烷(Tris)购自Biofroxx公司.
1.2 甲硫氨酸甲基选择性13C标记的细胞色素c的表达纯化
在超净台中将5 μL酿酒酵母iso-1细胞色素c质粒(-20 ℃冰箱保存)转入大肠杆菌BL21(DE3)感受态细胞(本实验室保存)中,混合均匀. 于冰上放置30 min,42 ℃水浴热激90 s,再在冰上放置1 min.加入500 μL无抗LB培养基,置于摇床(37 ℃,220 r/min)复苏培养30 min后,取150 μL涂于含氨苄青霉素的固体LB培养基上,待菌液干后,倒扣放入37 ℃恒温箱中培养8~9 h.挑取单菌落至装有5 mL LB培养基(加入5 μL 100 mg/mL氨苄青霉素)的离心管中,置于摇床(37 ℃,220 r/min)培养9~10 h.再按照菌液:培养基为1:100扩大培养:将离心管中的菌液离心(4 ℃,6 000 r/min)10 min,弃上清,用M9培养基重悬菌体,并将重悬菌液转入装有1 L M9培养基的锥形瓶(加入25 mg甲基13C标记的Met)中,置于摇床(37 ℃,220 r/min)培养至OD600 = 0.8时,加入100 μL母液为1 mmol/L的IPTG诱导剂,后再在1 L M9培养基补加75 mg甲基13C标记的Met.继续在30 ℃、70 r/min的条件下诱导表达蛋白质12~15 h,使用配备JA-10转子的贝克曼离心机(4 ℃,6 000 r/min)离心10 min,弃上清,收取湿菌体,待裂解纯化.
湿菌体重悬于50 mL裂解缓冲液(Lysis Buffer,含50 mmol/L Tris,5 mmol/L EDTA-2Na,pH 7.5),再加入溶菌酶(1.5 mg/g湿菌体)和5 μL脱氧核糖核酸酶I(DNase I),菌液在室温下消化1~2 h.使用高压破碎仪在800~1 000 MPa压力下破碎菌体15~20 min,使得菌液由浑浊变为透亮,获得大肠杆菌裂解液.再使用配备JA-25.5转子的贝克曼离心机(4 ℃,20 000 r/min)离心30 min.将离心后的上清倒入放有磁子的烧杯中,缓慢加入10.5 g固体硫酸铵并对溶液进行搅拌.待硫酸铵固体完全溶解后,将溶液放置在4 ℃冰箱中继续搅拌盐析3~6 h.盐析后,使用配备JA-25.5转子的贝克曼离心机(4 ℃,20 000 r/min)离心30 min. 将离心后的上清转入截流量为6 kDa透析袋中,透析袋放入5 L透析液中,在4 ℃冰箱中过夜透析. 透析结束后,使用配备JA-25.5转子的贝克曼离心机(4 ℃,20 000 r/min)离心30 min,取上清,用0.22 μm滤膜过滤,滤液待进一步分离纯化.
先用超纯水清洗纯化仪,后将Hitrap SP HP阳离子交换色谱柱(GE Healthcare)连接在纯化仪上,用超纯水冲洗阳离子交换柱至紫外吸收达到基线,再用Buffer A(20 mmol/L磷酸缓冲液,pH 7.0)冲洗平衡至紫外吸收基线稳定后上样. 上样结束后,先用Buffer A冲洗部分杂蛋白至紫外吸收线平稳,然后用20%~80%的Buffer B(20 mmol/L磷酸缓冲液,1 mol/L NaCl,pH 7.0)进行梯度洗脱,采用自动接样器以5 mL/管收集样品.使用3 kDa浓缩管将Buffer B浓度梯度大于40%的样品体积浓缩至5 mL以内,然后使用HiLoad Superdex 75 pg制备尺寸排阻色谱柱(GE Healthcare)进行纯化. 超纯水清洗色谱柱后,用Buffer C(20 mmol/L磷酸缓冲液,250 mmol/L NaCl,pH 7.0)对排阻色谱柱进行平衡,然后将样品加入上样环中使用Buffer C进行纯化. 同样使用自动接样器以5 mL/管收集样品,并根据280 nm紫外吸收峰(酿酒酵母iso-1细胞色素c的紫外吸收为430 760 L*mol-1*cm-1)来确定目标样品,取20 μL目标样品用于十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)和高分辨质谱(MS)分析.
纯化后样品经3 kDa浓缩管加入超纯水浓缩,冷冻干燥后存于-20 ℃冰箱待用.
1.3 圆二色谱实验
为了研究蛋白质的二级结构是否变化,我们对氧化修饰前后的Met甲基选择性13C标记的细胞色素c蛋白样品进行了圆二色谱实验.将Met甲基选择性13C标记的细胞色素c蛋白溶于磷酸钾缓冲液(20 mmol/L,pH 7.0),并添加不同浓度的H2O2,蛋白终浓度为25 μmol/L.
圆二色谱实验在Chirascan(Applied Photophysics)圆二色谱仪上进行.正式测样前,需要提前往检测器中通氮气30 min.实验中采集波长为180~260 nm范围内的圆二色谱,每次实验重复扫描三次后取平均值,并扣除相应的背景信号.通过Origin 9.1进行数据分析处理.
1.4 NMR实验
用于NMR实验的蛋白质样品溶于磷酸钾缓冲液(20 mmol/L,pH 7.0),并补加10% D2O用于锁场. 用于NMR实验的Met样品采用和蛋白相同的缓冲液.
所有NMR实验均在配备了TXI低温探头的Bruker Avance 700 MHz NMR谱仪上进行,实验温度为25 ℃.其中,二维1H-13C HSQC谱的F2(1H)和F1(13C)维的谱宽分别为14 094.74 Hz和7 042.83 Hz,中心分别为3 290.94 Hz和4 401.64 Hz,采样数据点阵为t2×t1 = 2 048×128,累加次数为16.在对Met末端甲基指认时,为了过滤Met中甲基以外其他相关信号,降低其对修饰甲基识别的影响,我们采用二维四量子滤波(1H-13C MAXY-HMQC)实验[24],通过选择性检测甲基来对Met甲基亚砜化前后的变化进行追踪.1H-13C MAXY-HMQC实验F2(1H)和F1(13C)维的谱宽分别为12 755.74 Hz和8 552.65 Hz,中心分别为3 290.94 Hz和4 401.64 Hz.采样数据点阵t2×t1 = 2 048×128,累加次数为8.谱图数据均采用Bruker TopSpin 4.0.1处理.
2 结果与讨论
2.1 甲硫氨酸甲基选择性13C标记的细胞色素c的表征
采用SDS-PAGE和MS对分离纯化的Met甲基选择性13C标记的细胞色素c蛋白的纯度进行检验. 如图1(a)所示,凝胶电泳图中只有一条蛋白质条带(cc),分子量接近12 kDa,说明蛋白质纯化后为单体且纯度较高.为了确定标记效率,我们对纯化后的样品进行了MS分子量测定,结果如图1(b)所示.细胞色素c氨基酸序列中一共含有三个Met,分别为Met-6、Met64和Met80.其中,Met-6为起始密码子对应的氨基酸,并不参与蛋白的主要功能;Met64被包埋于蛋白质结构内部,稳定性较强;Met80处于细胞色素c血红素基团附近,并与血红素形成配位,稳定性较差.三个Met末端甲基被13C标记后,蛋白质理论分子量由原来的12 666 Da变为12 669 Da,MS测得分子量为12 669.64 Da,与理论分析基本吻合,说明表达纯化的细胞色素c上所有Met甲基均成功获得13C标记.
图1
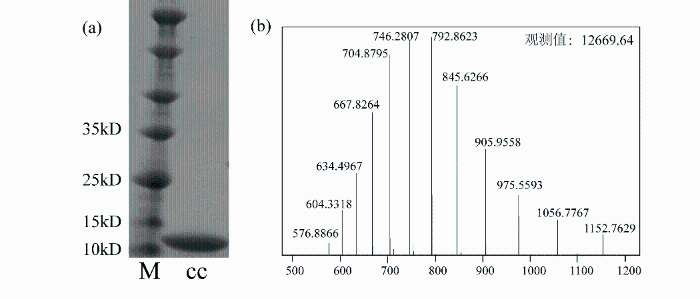
图1
纯化后的Met甲基选择性13C标记的细胞色素c检测. (a) SDS-PAGE. M为Maker,cc为Met甲基选择性13C标记的细胞色素c蛋白;(b)高分辨质谱
Fig. 1
Detection of methyl-13C methionine labeled cytochrome c after purification. (a) SDS-PAGE. M, Maker, cc, methyl-13C methionine labeled cytochrome c; (b) High-resolution mass spectrum
2.2 甲硫氨酸甲基选择性13C标记的细胞色素c的谱峰归属
细胞色素c是含铁原子的金属蛋白,通常有两种基本结构,即氧化态和还原态.当铁原子处于三价时,细胞色素c为氧化态,处于二价时则为还原态. 为了对氧化态和还原态的细胞色素c进行区分,我们通过外加铁氰化钾和抗坏血酸钠来调控细胞色素c的氧化态和还原态,并分别采集了依据文献[25]表达提纯13C全标记和Met甲基选择性13C标记的细胞色素c在不同状态下的1H-13C HSQC谱图.图2(a)和2(c)分别为加入十倍浓度(1 mmol/L)铁氰化钾和抗坏血酸钠的13C全标记细胞色素c的谱图,图2(b)和(d)分别为加入细胞色素c十倍浓度的铁氰化钾和抗坏血酸钠的Met甲基选择性13C标记的细胞色素c的谱图.从1H-13C HSQC谱图对比可明显看出,通过样品制备时采用选择性标记策略,使细胞色素c中仅有Met的甲基被13C标记,而其他氨基酸残基保持天然丰度,从而极大的简化了蛋白质的1H-13C HSQC二维谱图[20],减少了背景干扰,有利于对Met氧化修饰后信号进行分析指认.
图2
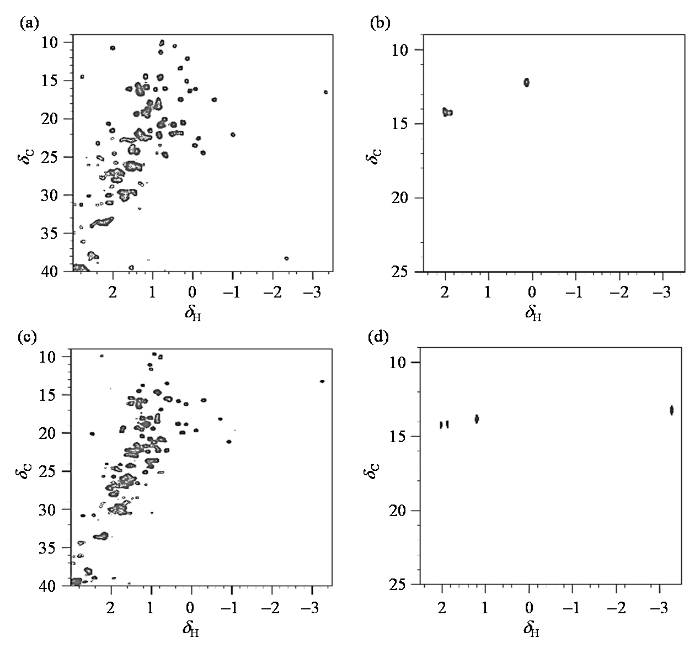
图2
不同状态下,13C全标记和Met甲基13C标记的细胞色素c的1H-13C HSQC谱图. (a) 0.1 mmol/L 13C全标记细胞色素c,氧化态;(b) 0.1 mmol/L Met甲基选择性13C标记细胞色素c,氧化态;(c) 0.1 mmol/L 13C全标记细胞色素c,还原态;(d) 0.1 mmol/L Met甲基选择性13C标记细胞色素c,还原态. (a)、(c)中Met-6相对信号强度较弱,在(b)、(d)中由于选择性标记了Met末端甲基,所以Met-6的信号明显
Fig. 2
1H-13C HSQC spectra of 13C full-labeled and methyl-13C methionine labeled cytochrome c under different states. (a) 0.1 mmol/L 13C full-labeled cytochrome c, oxidative state; (b) 0.1 mmol/L methyl- 13C labeling methione labeled cytochrome c, oxidative state; (c) 0.1 mmol/L 13C full-labeled cytochrome c, reductive state; (d) 0.1 mmol/L methyl- 13C methionine labeled cytochrome c, reductive state. (a)、(c) the signal intensity of Met-6 is weak. (b)、(d) the signal of Met-6 is obvious due to the selective labeling of methionine terminal methyl group
为了对不同状态下细胞色素c的Met分别进行指认,我们往0.1 mmol/L Met甲基选择性13C标记的细胞色素c样品中分别加入1 mmol/L(十倍浓度)的铁氰化钾或抗坏血酸钠来调控细胞色素c的氧化态和还原态. 图3(a)为0.1 mmol/L Met甲基选择性13C标记的细胞色素c的1H-13C HSQC谱图,存在较多信号,主要是由于此时既有氧化态,又有还原态.而加入1 mmol/L(十倍浓度)的铁氰化钾氧化后[图3(b)]或抗坏血酸钠还原后[图3(c)]的蛋白谱图明显得到了简化.铁氰化钾氧化后的谱图中仅有一个13C标记的Met信号存在,这主要是因为Met80被氧化后的信号由于Fe顺磁效应引发弛豫时间和化学位移变化,造成信号难以检测[25]. 而还原态有两个信号,分别对应于Met64和Met80.结合文献[26]中对细胞色素c的归属,我们可确定氧化态和还原态Met64的质子化学位移分别为δH 0.12和δH1.20,我们将其分别记为M64(ox)和M64(re).还原态Met80的化学位移为δH -3.30,被记为M80(re).需要指出的是,首位甲硫氨酸(Met-6,δH 2.05)位于蛋白质的N端易被切除,并不参与功能变化[27],因此我们在此对其未做详细讨论.
图3
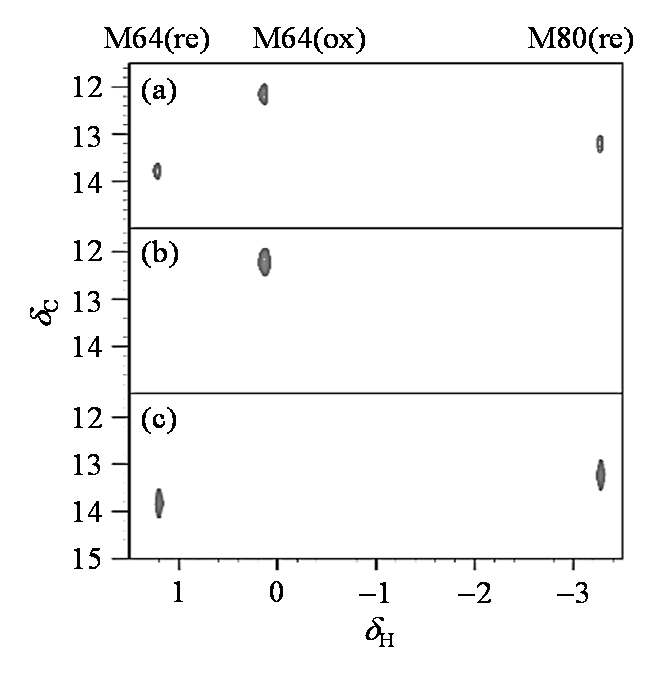
图3
不同状态的Met甲基13C标记的细胞色素c的1H-13C HSQC谱图. (a) 0.1 mmol/L Met甲基选择性13C标记的细胞色素c;(b)向(a)图样品中加入1 mmol/L(十倍浓度)铁氰化钾;(c)向(a)图样品中加入1 mmol/L(十倍浓度)抗坏血酸钠
Fig. 3
1H-13C HSQC spectra of methyl-13C methionine labeled cytochrome c under different states. (a) 0.1 mmol/L methyl- 13C methionine labeled cytochrome c; (b) Sample in Fig. (a) with addition of 1 mmol/L potassium ferricyanide; (c) Sample in Fig. (a) with addition of 1 mmol/L sodium ascorbate
2.3 不同氧化程度Met的信号归属
为了明确细胞色素c蛋白上Met氧化修饰后甲基信号,我们先用ChemDraw软件(ChemDraw20.0版)模拟了不同氧化修饰程度下游离Met末端甲基的化学位移(表1),并采集了游离的Met氧化修饰前后的NMR谱图,以便进行确认. Met氧化修饰后理论上会出现两种不同氧化程度的结果,一种为氧化程度较低的亚砜化修饰(硫原子上链接一个氧原子);另一种则为砜化,也是Met氧化程度最高的氧化修饰,在硫原子上连接了两个氧原子. 从表1模拟结果可以看到,一旦发生氧化修饰,末端甲基的1H和13C化学位移均向低场移动,其中13C化学位移远离常规的蛋白上甲基的化学位移范围,因此易于识别,可用于修饰信号的直接判断. 化学位移变化明显的主要原因在于与质子相连的碳原子上连接了电负性基团,由于吸电子效应,质子和碳原子周围电子云密度下降,屏蔽作用减弱,质子共振吸收移向低场,化学位移数值增大. 所以Met氧化修饰后甲基上的氢原子和碳原子的化学位移数值都比原始值有所增加.
表1 ChemDraw计算模拟的不同氧化程度Met末端甲基的化学位移
Table 1
| Molecules | δH of the terminal methyl | δC of the terminal methyl |
|---|---|---|
| Met | 2.07 | 15.4 |
| 亚砜化Met | 2.50 | 38.4 |
| 砜化Met | 2.80 | 43.8 |
为了验证模拟结果的准确性,我们采集了H2O2氧化前后游离的非标记Met的1H-13C MAXY-HMQC谱图.1H-13C MAXY-HMQC实验采用了四量子滤波,可选择性观测甲基的信号,追踪Met末端甲基的化学位移变化.如图4(a)所示,Met的1H-13C MAXY-HMQC谱图仅有一个信号存在,该信号显然来自其末端甲基.加入H2O2后[图4(b)],原来的甲基信号强度减弱,而低场区出现了新的信号. 该信号与ChemDraw软件预测的亚砜化修饰的Met末端甲基的化学位移接近,因此可以确定发生亚砜化修饰后的Met末端甲基信号化学位移远离未修饰的信号,处于(δH 2.65,δC 36.7)附近.
图4
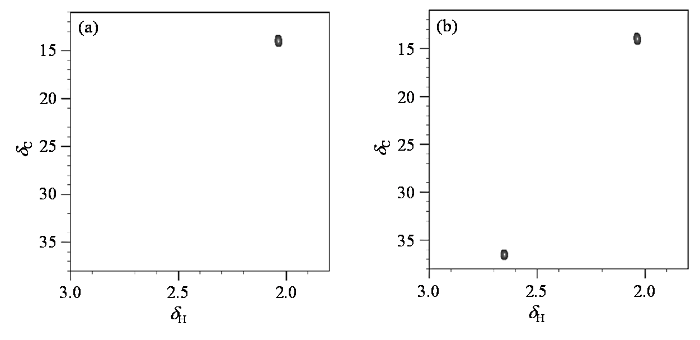
图4
游离的Met氧化前后的1H-13C MAXY-HMQC谱图. (a) 10 mmol/L Met;(b)向(a)图样品中加入20 mmol/L过氧化氢
Fig. 4
1H-13C MAXY-HMQC spectrum of free methionine before and after oxidation. (a) 10 mmol/L methionine; (b) Sample in Fig. (a) with addition of 20 mmol/L hydrogen peroxide
2.4 Met甲基选择性13C标记的细胞色素c的氧化过程观测
细胞色素c是一种多构象蛋白质,pH对其构象影响较大[28].为了减少碱式构象可能对氧化修饰观测过程的影响,我们选择滴加H2O2来观测该蛋白在活性氧存在时的氧化修饰过程. 将H2O2加入Met甲基选择性13C标记的细胞色素c后孵育30 min,再进行NMR谱图采集,分别按照细胞色素c:H2O2(cc:H2O2)为1:0、1:0.125、1:0.25、1:0.5、1:1、1:2、1:4、1:8的比例制备了8个样品.
因为细胞色素c已经选择性标记,所以使用1H-13C HSQC采样能节省采样时间且灵敏度更高. 不同浓度H2O2氧化处理的细胞色素c样品1H-13C HSQC谱图如图5所示. 图5(a)和5(b)分别为加入200 μmol/L(1:8的比例)前后细胞色素c的1H-13C HSQC谱图. 可以看出,当未添加H2O2时,氧化态和还原态信号均出现,说明初始状态下该蛋白氧化态和还原态共存. 但是氧化态Met64的信号远强于还原态Met64的信号,表明两态所占比例并非一致,氧化态蛋白所占比例高. 在高浓度活性氧条件下[图5(b)],蛋白的Met64和Met80的还原态信号均消失,但氧化态信号Met64(ox)和末端Met-6则无明显变化. 与之对应,在化学位移δH 2.60附近出现了新的信号,该信号1H和13C化学位移均与亚砜化修饰后游离Met的末端甲基化学位移接近,表明这一信号来自于亚砜化修饰甲基.
图5
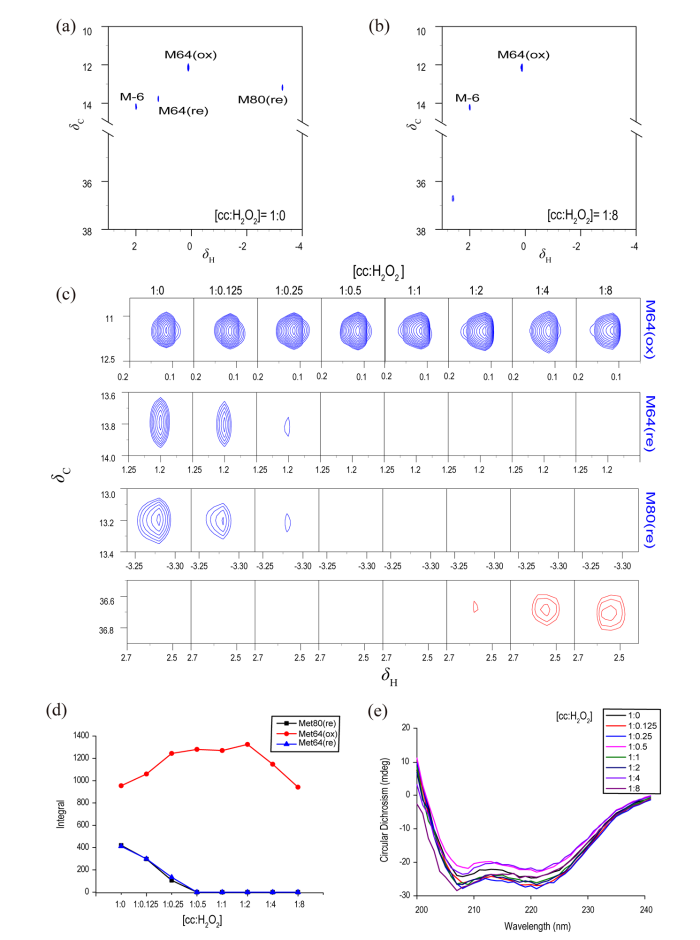
图5
不同氧化程度的Met甲基选择性13C标记细胞色素c的1H-13C HSQC和圆二色谱. (a) 25 μmol/L Met甲基选择性13C标记细胞色素的1H-13C HSQC谱;(b)向图(a)样品中加入200 μmol/L H2O2后的1H-13C HSQC谱;(c)不同浓度H2O2存在时,Met甲基选择性13C标记细胞色素c的1H-13C HSQC谱中不同Met甲基信号的变化;(d)不同浓度H2O2存在时,Met甲基选择性13C标记细胞色素c的不同Met甲基信号积分变化;(e)不同浓度H2O2存在时,Met甲基选择性13C标记细胞色素c的圆二色谱
Fig. 5
1H-13C HSQC spectra and CD spectra of methyl-13C methionine labeled cytochrome c in addition with hydrogen peroxide with different concentrations. (a) 1H-13C HSQC spectrum of 25 μmol/L methyl- 13C methionine labeled cytochrome c; (b) 1H-13C HSQC spectrum of sample in Fig. (a) in addition with 200 μmol/L H2O2; (c) Methyl signals in the 1H-13C HSQC spectra of methyl-13C methionine labeled cytochrome c in addition with different concentrations of hydrogen peroxide; (d) The methionine integrals of methyl-13C methionine labeled cytochrome c change with the concentrations of added hydrogen peroxide; (e) CD spectra of methyl-13C methionine labeled cytochrome c in addition with different concentrations of hydrogen peroxide
为了进一步明确其氧化修饰过程,我们对不同H2O2浓度下的相应基团信号进行了进一步分析,结果如图5(c)所示. 随着H2O2浓度的增加,不同信号的变化形式各不相同. 还原态的Met64(re)和Met80(re)的信号强度随过氧化氢浓度的增加而急剧减弱,表明两个还原态Met在氧化环境中所受的影响基本相似. 图5(d)为Met各信号的面积积分随H2O2浓度增加的变化情况. 其中氧化态Met64的积分强度有变化,说明在过氧化氢浓度增加到100 μmol/L(浓度比为1:4)时,可能存在少量Met64的继续氧化. 但是二维谱图中信号强弱变化与蛋白质的弛豫也有关联,使用其进行定量分析会引入误差.在过氧化氢浓度为12.5 μmol/L(浓度比为1:0.5)时,还原态Met64和Met80的信号完全消失不见,说明在此氧化环境中,细胞色素c完全转化为了氧化态. 但在此条件下,我们并没有观测到氧化修饰后亚砜化甲基信号的出现. 随着过氧化氢浓度的进一步增加,当蛋白与氧化剂的浓度比为1:2时,亚砜化甲基逐渐出现,且随着氧化剂浓度的增加信号强度逐步增强. 这说明在H2O2浓度增加过程中,细胞色素c的Fe优先被氧化,因而引发了该蛋白由还原态向氧化态转变,溶液中蛋白由氧化态和还原态两态并存逐渐统一为氧化态. 此后,活性氧的存在进一步引发蛋白发生氧化修饰. 细胞色素c受到赝化学位移效应的影响,Met80氧化修饰后谱线信号有化学位移扰动[29].
为了明确氧化修饰对细胞色素c蛋白结构的影响,我们进一步测定了不同H2O2浓度下的细胞色素c的圆二色谱. 结果如图5(e)所示,在氧化过程中,蛋白质的二级结构没有发生明显变化. 说明在H2O2低浓度时,蛋白的结构受影响较小. 有限的活性氧含量还不足以破坏细胞色素c的二级结构,主要对铁原子以及活性氧敏感的残基Met80起到氧化修饰作用.
实验显示,在调控氧化剂含量与蛋白质相当的氧化过程中,细胞色素c还原态逐渐信号消失,直至统一为氧化态,但在这个过程中Met氧化修饰信号并未立即出现. 此时细胞色素c可通过氧化还原对活性氧进行清除,维持动态平衡. 一方面说明了细胞色素c本身具有一定抵御活性氧的作用;另一方面也暗示了三价铁可能参与了蛋白质氧化修饰过程并起到催化的作用,这与Parakra等[30]的发现相符. 当H2O2含量在细胞色素c耐受范围内,细胞色素c只是在氧化态还原态之间进行转换,说明最先受到活性氧影响的是血红素基团中的铁原子,蛋白的结构和功能受到影响较小.
当H2O2浓度进一步增加,H2O2进入细胞色素c血红素疏水口袋,与血红素基团附近的Met80接触,从而引发Met80的氧化,而Met64却未被氧化. 而Bushnell等对马心细胞色素c的研究指出,除了Met80以外,马心细胞色素c的Met65也能够被氧化修饰[31],与我们实验结果有所差异,可能是两个蛋白结构不同所引发的. 如图6所示,酵母细胞色素c和马心细胞色素c晶体结构中Met80的位置基本重叠,但是Met64(红色,酵母)和Met65(粉色,马心)在蛋白结构中的位置却存在明显差异. 酵母细胞色素c的Met64被包埋在蛋白质内部,通常情况下难以被氧化,这与Berghuis等[32]的发现相符. 而马心细胞色素c的Met65位于蛋白质表面,易于接触活性氧,因此易被氧化. 我们的发现说明不同来源的细胞色素c因为在结构上存在差异,所以即使在相同条件下,氧化修饰的程度和位点也会存在差异. 因此不同来源的细胞色素c很多功能(如过氧化物酶活性)存在较大差别. 此外,研究中发现Met80发生亚砜化氧化修饰后,蛋白的结构变化不大. 一方面说明此时可能只是部分蛋白质发生氧化修饰;另一方面表明Met过度氧化或者其他残基发生修饰后的协同作用才可引发细胞色素c的结构变化,使其发挥较强的过氧化物酶活性,这与Yin等[19]的报道相符. 综上所述,本研究发现在活性氧作用初期,酵母细胞色素c存在一种被部分氧化修饰但又保持整体结构稳定的状态,暗示着活性氧诱导的细胞色素c氧化可能历经多个阶段,这对进一步理解细胞色素c在氧化应激条件下的调控功能具有重要意义.
图6
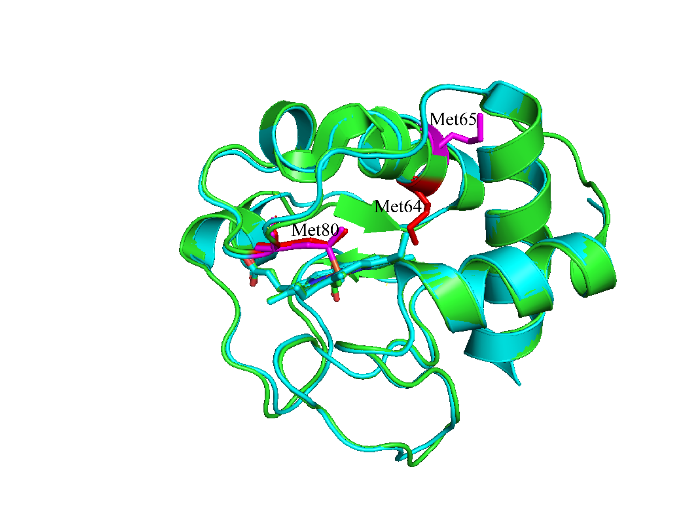
图6
酵母细胞色素c(绿色,PDB:1YCC)和马心细胞色素c(蓝色,PDB:1HRC)结构图
Fig. 6
The structure of yeast cytochrome c (green, PDB: 1YCC) and horse heart cytochrome c (blue, PDB: 1HRC)
3 结论
为揭示活性氧含量对于细胞色素c功能转变的调控作用,本研究通过对酵母细胞色素c上Met末端甲基进行13C标记,对不同浓度H2O2存在时的细胞色素c上Met的氧化修饰情况进行了观测. 研究发现,随着H2O2含量的增加,酵母细胞色素c蛋白先由氧化态和还原态两态并存逐渐转化为全氧化态. 当H2O2浓度进一步增加时,Met才发生氧化修饰,且该氧化主要发生在Met80上. 结合圆二色谱实验发现蛋白Met的亚砜化修饰后,对细胞色素c的二级结构影响很小,并没有导致明显的变化. 以上结果暗示着细胞色素c在细胞内活性氧含量增加时,具有一定抵御活性氧的作用;只有当活性氧含量持续增加,细胞色素c上Met才会发生氧化修饰.说明了活性氧对细胞色素c的翻译后修饰可能需要多个基团参与,才可对其结构和功能产生调控.
利益冲突
无
致谢
感谢国家重点基础研究发展计划的支持(2017YFA0505400,2018YFE0202300,2018YFA0704002).
参考文献
Role of metabolic H2O2 generation: redox signaling and oxidative stress
[J].DOI:10.1074/jbc.R113.544635 URL [本文引用: 1]
LC-MS/MS suggests that hole hopping in cytochrome c peroxidase protects its heme from oxidative modification by excess H2O2
[J].DOI:10.1039/C6SC03125K URL [本文引用: 1]
Ligation and reactivity of methionine-oxidized cytochrome c
[J].
DOI:10.1021/acs.inorgchem.8b00010
PMID:29708337
[本文引用: 2]

Met80, one of the heme iron ligands in cytochrome c (cyt c), is readily oxidized to Met sulfoxide (Met-SO) by several biologically relevant oxidants. The modification has been suggested to affect both the electron-transfer (ET) and apoptotic functions of this metalloprotein. The coordination of the heme iron in Met-oxidized cyt c (Met-SO cyt c) is critical for both of these functions but has remained poorly defined. We present electronic absorption, NMR, and EPR spectroscopic investigations as well as kinetic studies and mutational analyses to identify the heme iron ligands in yeast iso-1 Met-SO cyt c. Similar to the alkaline form of native cyt c, Lys73 and Lys79 ligate to the ferric heme iron in the Met80-oxidized protein, but this coordination takes place at much lower pH. The ferrous heme iron is ligated by Met-SO, implying the redox-linked ligand switch in the modified protein. Binding studies with the model peptide microperoxidase-8 provide a rationale for alterations in ligation and for the role of the polypeptide packing in native and Met-SO cyt c. Imidazole binding experiments have revealed that Lys dissociation from the ferric heme in K73A/K79G/M80K (M80K) and Met-SO is more than 3 orders of magnitude slower than the opening of the heme pocket that limits Met80 replacement in native cyt c. The Lys-to-Met-SO ligand substitution gates ET of ferric Met-SO cyt c with Co(terpy). Owing to the slow Lys dissociation step, ET reaction is slow but possible, which is not the case for nonswitchable M80A and M80K. Acidic conditions cause Lys replacement by a water ligand in Met-SO cyt c (p K = 6.3 ± 0.1), increasing the intrinsic peroxidase activity of the protein. This pH-driven ligand switch may be a mechanism to boost peroxidase function of cyt c specifically in apoptotic cells.
Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide-sequences
[J].DOI:10.1002/(ISSN)1097-0134 URL [本文引用: 1]
Methionine in proteins defends against oxidative stress
[J].
DOI:10.1096/fj.08-118414
PMID:18845767
[本文引用: 1]
A variety of reactive oxygen species react readily with methionine residues in proteins to form methionine sulfoxide, thus scavenging the reactive species. Most cells contain methionine sulfoxide reductases, which catalyze a thioredoxin-dependent reduction of methionine sulfoxide back to methionine. Thus, methionine residues may act as catalytic antioxidants, protecting both the protein where they are located and other macromolecules. To test this hypothesis directly, we replaced 40% of the methionine residues in Escherichia coli with norleucine, the carbon-containing analog, in which the sulfur of methionine is substituted by a methylene group (-CH2-). The intracellular free methionine and S-adenosylmethionine pools were not altered, nor was the specific activity of the key enzyme, glutamine synthetase. When unstressed, both control and norleucine-substituted cells survived equally well at stationary phase for at least 32 h. However, oxidative stress was more damaging to the norleucine-substituted cells. They died more rapidly than control cells when challenged by hypochlorite, hydrogen peroxide, or ionizing radiation. One of the most abundant proteins in the cell, elongation factor Tu, was found to be more oxidatively modified in norleucine-substituted cells, consistent with loss of the antioxidant defense provided by methionine residues. The results of these studies support the hypothesis that methionine in protein acts as an endogenous antioxidant in cells.
Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems
[J].Aging and some pathological conditions are associated with the accumulation of altered (inactive or less active) forms of enzymes. It was suggested that these age-related alterations reflect spontaneous changes in protein conformation and/or posttranslational modifications (e.g., oxidation). Because changes in protein conformations are often associated with changes in surface hydrophobicity, we have examined the effects of aging and oxygen radical-dependent oxidation on the hydrophobicity of rat liver proteins. As a measure of hydrophobicity, the increase in fluorescence associated with the binding of 8-anilino-1-naphthalene-sulfonic acid to hydrophobic regions on the proteins was used. By this criterion, the hydrophobicity of liver proteins of 24-month-old rats was 15% greater than that of 2-month-old animals. Exposure of liver proteins to a metal-catalyzed oxidation system (ascorbate/Fe(II)/H2O2) or a peroxyl radical generating system, 2,2'-azobis(2-amidinopropane) dihydrochloride (AAPH) led to increases of 2% or 30% in surface hydrophobicity, respectively. Treatment of liver proteins with the metal-catalyzed oxidation system led to a significant increase in reactive carbonyl content and to conversion of methionine residues to methionine sulfoxide residues. Treatment with AAPH led also to oxidation of methionine, tyrosine, and tryptophan residues and to the precipitation of some proteins. Dityrosine was detected in AAPH-treated protein, both the precipitate and supernatant fraction. The oxidation-dependent increase of hydrophobicity was correlated with an increase in the levels of methionine sulfoxide and dityrosine. These results suggest that oxidative modification of proteins may be responsible for the age-related increase of protein surface hydrophobicity in vivo, and that the oxidation of methionine by an oxidative system may be an important event for the change of protein conformation.
The ubiquitin pathway for the degradation of intracellular proteins
[J].
Protein oxidation-formation mechanisms, detection and relevance as biomarkers in human diseases
[J].DOI:10.1016/j.redox.2021.101901 URL [本文引用: 1]
Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c
[J].
DOI:10.1016/s0092-8674(00)80085-9
PMID:8689682
[本文引用: 2]
A cell-free system based on cytosols of normally growing cells is established that reproduces aspects of the apoptotic program in vitro. The apoptotic program is initiated by addition of dATP. Fractionation of cytosol yielded a 15 kDa protein that is required for in vitro apoptosis. The absorption spectrum and protein sequence revealed that this protein is cytochrome c. Elimination of cytochrome c from cytosol by immunodepletion, or inclusion of sucrose to stabilize mitochondria during cytosol preparation, diminished the apoptotic activity. Adding back cytochrome c to the cytochrome c-depleted extracts restored their apoptotic activity. Cells undergoing apoptosis in vivo showed increased release of cytochrome c to their cytosol, suggesting that mitochondria may function in apoptosis by releasing cytochrome c.
Multifunctional cytochrome c: learning new tricks from an old dog
[J].DOI:10.1021/acs.chemrev.7b00257 URL [本文引用: 1]
Post-translational modifications of cytochrome c in cell life and Disease
[J].
DOI:10.3390/ijms21228483
URL
[本文引用: 1]
Mitochondria are the powerhouses of the cell, whilst their malfunction is related to several human pathologies, including neurodegenerative diseases, cardiovascular diseases, and various types of cancer. In mitochondrial metabolism, cytochrome c is a small soluble heme protein that acts as an essential redox carrier in the respiratory electron transport chain. However, cytochrome c is likewise an essential protein in the cytoplasm acting as an activator of programmed cell death. Such a dual role of cytochrome c in cell life and death is indeed fine-regulated by a wide variety of protein post-translational modifications. In this work, we show how these modifications can alter cytochrome c structure and functionality, thus emerging as a control mechanism of cell metabolism but also as a key element in development and prevention of pathologies.
Cytochrome c: An extreme multifunctional protein with a key role in cell fate
[J].
Oxidative modification of cytochrome c by singlet oxygen
[J].DOI:10.1016/j.freeradbiomed.2007.12.031 URL [本文引用: 1]
Self-oxidation of cytochrome c at methionine80 with molecular oxygen induced by cleavage of the Met-heme iron bond
[J].
DOI:10.1039/c4mb00285g
PMID:25224641
[本文引用: 1]
Met80 of cytochrome c (cyt c) has been shown to dissociate from its heme iron when cyt c interacts with cardiolipin (CL), which triggers the release of cyt c into the cytosol initiating apoptosis. We found that the mass of human cyt c increases by 16 Da in the Met80-Lys86 region by reaction with molecular oxygen in the presence of CL-containing liposomes and dithiothreitol (DTT). To investigate the effect of Met80 dissociation on the reaction of cyt c with molecular oxygen without affecting its secondary structures, a human cyt c mutant (Δ8384 cyt c) was constructed by removing two amino acids (Val83 and Gly84) from the loop containing Met80. According to MALDI-TOF-MS and tandem mass measurements, Met80 of Δ8384 cyt c was modified site-specifically to methionine sulfoxide when purified in the presence of molecular oxygen, whereas Met80 was not modified in the absence of molecular oxygen. A red-shift of the Soret band from 406 to 412 nm and absorption increase at ∼536 and ∼568 nm were observed for Δ8384 cyt c when it reacted with DTT and molecular oxygen, followed by a further red-shift of the Soret band to 416 nm and absorption increase at ∼620 and ∼650 nm. These results indicate that Met80 of cyt c is oxidized site-specifically by formation of the oxy and subsequent compound I-like species when Met80 dissociates from the heme iron, where the Met80 modification may affect its peroxidase activity related to apoptosis.
Methionine sulfoxide cytochrome-c
[J].DOI:10.1021/bi00650a029 URL [本文引用: 1]
Locked and loaded for apoptosis
[J].DOI:10.1126/science.aan5587 PMID:28642398 [本文引用: 1]
Structural transformations of cytochrome c upon interaction with cardiolipin
[J].
Early modification of cytochrome c by hydrogen peroxide triggers its fast degradation
[J].
Lysine carbonylation is a previously unrecognized contributor to peroxidase activation of cytochrome c by chloramine-T
[J].DOI:10.1039/C8SC03624A URL [本文引用: 2]
NMR studies of large protein dynamics using unnatural amino acids
[J].
非天然氨基酸在蛋白质动态特性核磁共振研究中的应用
[J].
Methyl TROSY spectroscopy: a versatile NMR approach to study challenging biological systems
[J].
An integrated approach to unique NMR assignment of methionine methyl resonances in proteins
[J].
DOI:10.1021/acs.analchem.6b03705
PMID:28208280
[本文引用: 1]
The application of methyl nuclear magnetic resonance (NMR) spectroscopy in protein side-chain structural studies offers unique advantages of greater peak sensitivity, even for high-molecular-weight proteins. Traditionally, the utility of methyl NMR has often been limited by the difficulty in assigning the methyl resonances. Herein, a mass spectrometry (MS)-assisted strategy to assign the methyl resonances of methionine residues is presented. The strategy involves partially oxidizing the methionine and quantifying the oxidation level by both NMR and liquid chromatography-mass spectrometry (LC-MS). The NMR assignment of methyl resonances of methionine is made by correlating the quantitative results obtained from both NMR and MS. The method has been successfully demonstrated using the proteins hen egg-white lysozyme (HEWL) and porcine pepsin. The technique described herein can help facilitate the application of methyl NMR as a useful tool to study protein structure, dynamics, and interactions.
Evolution of amino acid frequencies in proteins over deep time: Inferred order of introduction of amino acids into the genetic code
[J].To understand more fully how amino acid composition of proteins has changed over the course of evolution, a method has been developed for estimating the composition of proteins in an ancestral genome. Estimates are based upon the composition of conserved residues in descendant sequences and empirical knowledge of the relative probability of conservation of various amino acids. Simulations are used to model and correct for errors in the estimates. The method was used to infer the amino acid composition of a large protein set in the Last Universal Ancestor (LUA) of all extant species. Relative to the modern protein set, LUA proteins were found to be generally richer in those amino acids that are believed to have been most abundant in the prebiotic environment and poorer in those amino acids that are believed to have been unavailable or scarce. It is proposed that the inferred amino acid composition of proteins in the LUA probably reflects historical events in the establishment of the genetic code.
Observation of separate J-resolved 1H NMR spectra from CH, CH2, and CH3 groups using a maximum-quantum filter
[J].
Conformational change of wild type cytochrome c characterized by NMR spectroscopy at natural isotropic abundance
[J].
天然同位素丰度野生型酵母细胞色素c构象变化的核磁共振检测
[J].
13C and proton NMR studies of horse cytochrome c assignment and temperature dependence of methyl resonances
[J].DOI:10.1016/0014-5793(86)80054-0 URL [本文引用: 1]
Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid
[J].In a significant fraction of the Escherichia coli cytosolic proteins, the N-terminal methionine residue incorporated during the translation initiation step is excised. The N-terminal methionine excision is catalyzed by methionyl-aminopeptidase (MAP). Previous studies have suggested that the action of this enzyme could depend mainly on the nature of the second amino acid residue in the polypeptide chain. In this study, to achieve a systematic analysis of the specificity of MAP action, each of the 20 amino acids was introduced at the penultimate position of methionyl-tRNA synthetase of E. coli and the extent of in vivo methionine excision was measured. To facilitate variant protein purification and N-terminal sequence determination, an expression shuttle vector based on protein fusion with beta-galactosidase was used. From our results, methionine excision catalyzed by MAP is shown to obey the following rule: the catalytic efficiency of MAP, and therefore the extent of cleavage, decreases in parallel with the increasing of the maximal side-chain length of the amino acid in the penultimate position. This molecular model accounts for the rate of N-terminal methionine excision in E. coli, as deduced from the analysis of 100 protein N-terminal sequences.
Monitoring alkaline transitions of yeast iso-1 cytochrome c at natural isotopic abundance using trimethyllysine as a native NMR probe
[J].
DOI:10.1039/c8cc07605g
PMID:30351312
[本文引用: 1]
Spectral overlap makes it difficult to use NMR for mapping the conformational profile of heterogeneous conformational ensembles of macromolecules. Here, we apply a 1H-14N HSQC experiment to monitor the alkaline conformational transitions of yeast iso-1 cytochrome c (ycyt c) at natural isotopic abundance. Trimethylated Lys72 of ycyt c is selectively detected by a 1H-14N HSQC experiment, and used as a probe to trace conformational transitions of ycyt c under alkaline conditions. It was found that at least four different conformers of ycyt c coexisted under alkaline conditions. Besides the native structure, Lys73 or Lys79 coordinated conformers and a partially unfolded state with exposed heme were observed. These results indicate that the method is powerful at simplifying spectra of a trimethylated protein, which makes it possible to study complex conformational transitions of naturally extracted or chemically modified trimethylated protein at natural isotopic abundance.
Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR
[J].The physiological complex of yeast cytochrome c peroxidase and iso-1-cytochrome c is a paradigm for biological electron transfer. Using paramagnetic NMR spectroscopy, we have determined the conformation of the protein complex in solution, which is shown to be very similar to that observed in the crystal structure [Pelletier H, Kraut J (1992) Science 258:1748-1755]. Our results support the view that this transient electron transfer complex is dynamic. The solution structure represents the dominant protein-protein orientation, which, according to our estimates, is occupied for >70% of the lifetime of the complex, with the rest of the time spent in the dynamic encounter state. Based on the observed paramagnetic effects, we have delineated the conformational space sampled by the protein molecules during the dynamic part of the interaction, providing experimental support for the theoretical predictions of the classical Brownian dynamics study [Northrup SH, Boles JO, Reynolds JCL (1988) Science 241:67-70]. Our findings corroborate the dynamic behavior of this complex and offer an insight into the mechanism of the protein complex formation in solution.
The proportion of Met80-sulfoxide dictates peroxidase activity of human cytochrome c
[J].
DOI:10.1039/c8dt02185f
PMID:29944150
[本文引用: 1]
The peroxidase activity of cytochrome c is proposed to contribute to apoptosis by peroxidation of cardiolipin in the mitochondrial inner membrane. However, cytochrome c heme is hexa-coordinate with a methionine (Met80) on the distal side, stopping it from acting as an efficient peroxidase. The first naturally occurring variant of cytochrome c discovered, G41S, has higher peroxidase activity than wild-type. To understand the basis for this increase and gain insight into the peroxidase activity of wild-type, we have studied wild-type, G41S and the unnatural variant G41T. Through a combined kinetic and mass spectrometric analysis, we have shown that hydrogen peroxide specifically oxidizes Met80 to the sulfoxide. In the absence of substrate this can be further oxidized to the sulfone, leading to a decrease in peroxidase activity. Peroxidase activity can be correlated with the proportion of sulfoxide present and if fully in that form, all variants have the same activity without a lag phase caused by activation of the protein.
Oxidation state-dependent conformational changes in cytochrome c
[J].
High-resolution 3-dimensional structure of horse heart cytochrome c
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}