


引言
瞬时相互作用的产生伴随着遭遇复合物的形成,遭遇复合物系综结构通过在方向集合中快速交换帮助蛋白质寻找结合位点[7].遭遇复合物通过增加相互作用横截面以及减少特异性复合物形成路径上的构象搜索空间来提高结合速率[8⇓⇓⇓⇓-13].尽管遭遇复合物在蛋白质结合中起到重要作用,但由于遭遇复合物是由弱相互作用驱动产生[14],其在溶液体系中丰度低、停留时间短,因此常规的结构生物方法,如X射线晶体衍射(X-ray Diffraction by Crystals,X-ray)、冷冻电镜(Cryo-Electron Microscopy,cryo-EM)等,很难有效地捕获其结构信息.核磁共振(Nuclear Magnetic Resonance,NMR)技术可以通过检测蛋白质复合物中主链酰胺键的化学位移变化来解析遭遇复合物的动态结构.在遭遇复合物中,蛋白质被溶剂化,酰胺键的化学环境变化小,化学位移扰动很小.而在特异性复合物中,溶剂层变化剧烈,酰胺键扰动较大[15⇓-17].其中,顺磁弛豫增强(Paramagnetic Relaxation Enhancement,PRE)已被证明是一种研究遭遇复合物结构的有效技术,其原理为顺磁中心的未配对电子产生的顺磁效应改变了附近原子核的共振频率及弛豫速率[18].此外,伪接触位移(Pseudocontact Shifts,PCS)[19]和残余偶极耦合(Residual Dipolar Couplings,RDC)[20,21]也可用于表征遭遇复合物动态结构.分子动力学(Molecular Dynamics,MD)模拟能够在毫秒时间尺度内观察分子运动,并在原子水平上提供动力学运动细节,因而被广泛用于蛋白质结合的动力学及热力学信息计算[22⇓⇓-25]. 由于模拟数据的不确定性,通常需要在多平行轨道上对蛋白质间相互作用进行长时间模拟[26⇓⇓⇓⇓-31].目前,遭遇复合物结构及其动力学信息已可以通过MD模拟进行合理预测,并与实验结果进行对比.
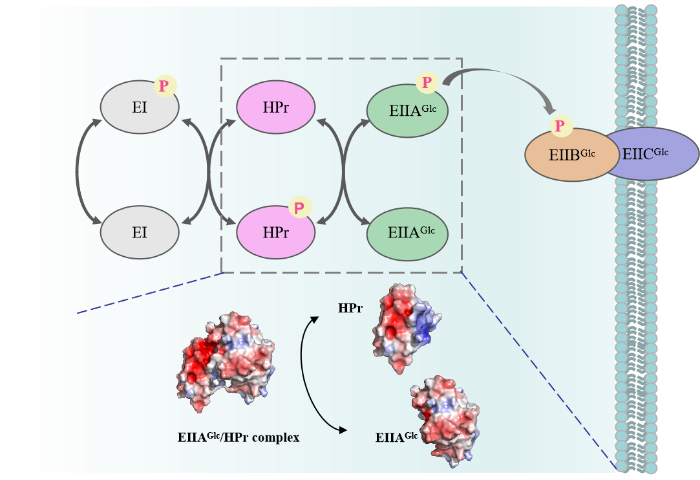
作为一种常见的磷酸信号网络系统,糖磷酸转移酶系统(Sugar Phosphotransferase System,PTS)参与介导碳水化合物的摄取、磷酸化及信号传导过程(图1).PTS主要由酶I(EI)、组氨酸磷酸载体蛋白(Histidine Phosphate Carrier Protein,HPr)和酶II(EIIAGlc)组成,通过分子间相互作用传递磷酸根[32].HPr与EIIAGlc的晶体结构及其特异性复合物结构已通过X-ray与NMR技术解析出来[33-34]. 此后,PRE与化学交联质谱技术的发展也使得EI/EIIAGlc、EI/HPr的相互作用及其调控机制依次得以研究[1,35⇓-37]. 但HPr与EIIAGlc的遭遇复合物结构及其作用机制仍不明确.本文选用PTS中的EIIAGlc/HPr复合物作为研究模型,结合PRE技术与MD模拟选择性检测EIIAGlc/HPr遭遇复合物中HPr的结构信息,我们发现在EIIAGlc/HPr特异性复合物形成过程中,HPr首先在扩散作用驱动下分布在EIIAGlc周围形成三种不同取向的遭遇复合物,之后不断向特异性复合物结合界面移动,与EIIAGlc形成特异性相互作用.
图1
1 实验部分
1.1 仪器与试剂
三羟甲基氨基甲烷(Tris)、异丙基-β-D-硫代半乳糖苷(IPTG)购自广州赛国生物科技有限公司;D-glucose、氯化钠(NaCl)、氯化镁(MgCl2)、磷酸二氢钾(KH2PO4)、十二水合磷酸氢二钠(Na2HPO4•12H2O)、葡萄糖购自国药集团化学试剂有限公司;酵母粉(Yeast extract)、胰蛋白胨(Tryptone)购自Oxoid公司;二硫苏糖醇(DTT)购自北京博奥拓达科技有限公司;N-15标记的氯化铵(N-15H4Cl)购自Sigma-Aldrich;乙二胺四乙酸(EDTA,cat#P996250)购自多伦多研究化学品公司;二甲基亚砜(DMSO)购自生工生物工程(上海)股份有限公司.
1.2 质粒构建、蛋白表达和纯化
将编码HPr、EIIAGlc的基因克隆到pET11a表达载体中.采用QuikChange[39]方法对HPr基因进行E5C和E66C半胱氨酸突变.大肠杆菌(BL21 star)细胞用于蛋白质表达,在LB和M9培养基中分别制备未标记的野生型HPr、HPr E5C、HPr E66C蛋白和N-15同位素标记的EIIAGlc蛋白.用阴离子交换柱和Sephacryl S100色谱柱(GE Healthcare)对这4种蛋白进行纯化.纯化后的蛋白用变性胶电泳和电喷雾-四极杆-飞行时间质谱仪(Agilent6530)进行分子量鉴定,并在Tris缓冲液(20 mmol/L Tris,150 mmol/L NaCl,pH 7.4)中保存.
用EDTA-Mn2+探针连接未标记的HPr E5C、HPr E66C蛋白,以制备EDTA-Mn2+ HPr E5C、EDTA-Mn2+ HPr E66C:首先在DMSO体系中将EDTA与两倍浓度的氯化锰混合制备EDTA-Mn2+探针;然后将制备好的探针分别与HPr E5C、HPr E66C蛋白以4:1摩尔比例混合,室温反应4 h得到EDTA-Mn2+ HPr E5C、 EDTA-Mn2+ HPr E66C蛋白;最后使用阴离子交换柱提纯连接成功的EDTA-Mn2+ HPr E5C、EDTA-Mn2+ HPr E66C蛋白,采用Amicon管浓缩并置换至最终实验缓冲液(20 mmol/L Tris,150 mmol/L NaCl,pH 7.4)中,此时溶液体系中的Mn2+离子已被去除.使用电喷雾-四极杆-飞行时间质谱仪(Agilent6530)进行分子量鉴定.
将浓度为300 μmol/L的N-15同位素标记的EIIAGlc蛋白与等浓度的未标记野生型HPr、HPr E5C、HPr E66C蛋白分别混合,制备野生型EIIAGlc/HPr、EIIAGlc/HPr E5C和EIIAGlc/HPr E66C复合物抗磁样品用于HSQC及PRE实验.样品缓冲液体系含20 mmol/L Tris、150 mmol/L NaCl、5% D2O,pH 7.4.
将浓度为300 μmol/L的N-15同位素标记的EIIAGlc蛋白与等浓度的未标记EDTA-Mn2+ HPr E5C、EDTA-Mn2+ HPr E66C蛋白分别混合,制备EIIAGlc/EDTA-Mn2+ HPr E5C和EIIAGlc/EDTA-Mn2+ HPr E66C复合物顺磁样品用于PRE实验.样品缓冲液体系含20 mmol/L Tris、150 mmol/L NaCl、5% D2O,pH 7.4.
1.3 NMR实验及数据处理
为确定半胱氨酸的引入不会干扰EIIAGlc/HPr的天然结构,对EIIAGlc/HPr E5C和EIIAGlc/HPr E66C分别采集1H-15N HSQC图谱,并与野生型EIIAGlc/HPr的谱图进行比较.1H-15N HSQC谱图由配备QCI超低温探头的Bruker 600 MHz NMR谱仪在298 K采集.F2维谱宽为16 ppm,F1维谱宽为30 ppm,采样点数t2×t1=2 048×256,所得1H-15N HSQC数据用NMRPipe(version 2010.160.15.01)处理.
基于1H-15N HSQC改进的氢核横向弛豫测定序列1hr2_hsqc_wgfb.ct,对EIIAGlc/HPr E5C、EIIAGlc/HPr E66C采集抗磁PRE图谱,对EIIAGlc/EDTA-Mn2+ HPr E5C和EIIAGlc/EDTA-Mn2+ HPr E66C采集顺磁PRE图谱.将同种突变的抗磁PRE结果与顺磁PRE结果进行对比获得蛋白质复合物的弛豫增强速率.PRE实验在配备QCI超低温探头的Bruker 600 MHz NMR谱仪上,298 K下采用两点法进行,两个时间点间的时间延迟为10 ms,所得实验数据用NMRPipe进行处理.
1.4 复合物建模与分子模拟
根据EIIAGlc/HPr E5C和EIIAGlc/HPr E66C遭遇复合物的PRE计算结构对HPr的空间分布进行方向簇分类,每个方向簇取一个代表性结构,将探针删除并还原后得到野生型EIIAGlc/HPr遭遇复合物结构.对EIIAGlc/HPr遭遇复合物及EIIAGlc/HPr特异性复合物结构(PDB:1GGR),在Amber 14中在ff14SB力场下进行MD模拟.整个模拟过程的温度为25℃,EIIAGlc/HPr复合物溶解在一个含有TIP3P水分子的周期盒中,每个方向与周期盒边界的距离至少为10 Å(1 Å=0.1 nm).模拟时间为200 ns,以100 ps为间隔捕捉2 000帧结构.每个实验对象以不同的随机数种子运行3条独立的MD轨道.最后使用AMBER 14中的CPPTRAJ模块计算每帧中遭遇复合物的1~235号残基的Cα均方根偏差(Root Mean Square Deviation,RMSD)及质心间距离值.
2 结果与讨论
2.1 EIIAGlc/HPr复合物系统中存在多种瞬时结构
PRE实验通常以主链酰胺键的氢核为观察核,向蛋白质复合物体系中引入顺磁性探针以引起自旋核的弛豫速率增强.由于主链酰胺键氢核的横向弛豫增强速率(
其中,
图2
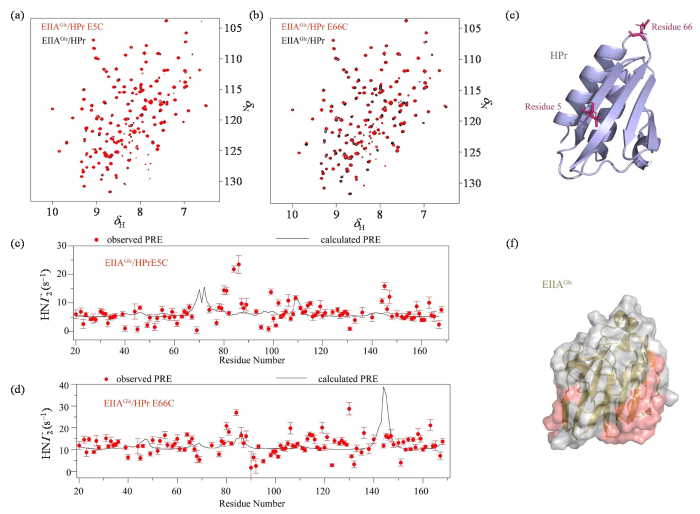
图2
25 ℃下,EIIAGlc/HPr溶液体系(黑色)与EIIAGlc/HPr E5C (a)、EIIAGlc/HPr E66C (b)溶液体系(红色)的1H-15N HSQC谱图比较;EIIAGlc与HPr E5C (c)、EIIAGlc与HPr E66C (d)的分子间PRE实验观测值(红色圆点)和理论计算值(黑色实线);(e) HPr探针连接位点结构图;(f) EIIAGlc的结构示意图,其中PRE实验值与理论计算值差异较大的区域用红色表示
Fig. 2
1H-15N HSQC spectra of EIIAGlc/HPr solution (black) and EIIAGlc/HPr E5C (a) or EIIAGlc/HPr E66C (b) (red) were compared under 25 ℃. Experimental observations (red dots) and theoretical calculations (black solid lines) of intermolecular PRE of EIIAGlc and HPr E5C (c) or HPr E66C (d); (e) Structure diagram of HPr probe connection site; (f) Schematic diagram of EIIAGlc structure, where areas with large differences between PRE experimental values and theoretical calculated values are shown in red
然而,我们发现EIIAGlc/HPr E5C、EIIAGlc/HPr E66C两种复合物中均有一些残基的PRE实验观测值与理论计算值相差较大[图2(c), (d), (f)],如EIIAGlc/HPr E5C复合物中EIIAGlc的63~85、140~145号残基,EIIAGlc/HPr E66C复合物中EIIAGlc的80~85、110~120、140~145号残基.这说明EIIAGlc/HPr E5C、EIIAGlc/HPr E66C蛋白质复合体不是以单一的特异性复合物形式存在于溶液中,EIIAGlc与HPr突变体蛋白之间还存在瞬时相互作用.因为各个EIIAGlc/HPr复合物的动态结构各不相同,而图2中的PRE理论计算基于单个特异性复合物的结构信息进行,PRE图谱反映的则是溶液体系中蛋白质复合物所有平衡状态的平均值,复合物体系中不同构象之间的交换通常比NMR的时间尺度快,故实验值与数据值之间的数值偏差可以证明溶液体系中蛋白质系综结构的存在.
2.2 EIIAGlc/HPr复合物系综结构的理论计算
我们通过分子对接方法生成含有3 600个EIIAGlc/HPr复合物的结构库,对构象库中每一个EIIAGlc/HPr复合物结构进行PRE理论值计算后与其实验值进行比较.由图3(a)知,EIIAGlc/HPr E5C和EIIAGlc/HPr E66C复合物系综结构中HPr的空间分布大体相同.对于两种复合物系综结构的PRE理论计算值与实验值的相关性,本文使用皮尔逊相关系数(Pearson correlation coefficient)进行度量. 皮尔逊相关系数(r)为两个变量之间的协方差与标准差的商,表示为:
图3
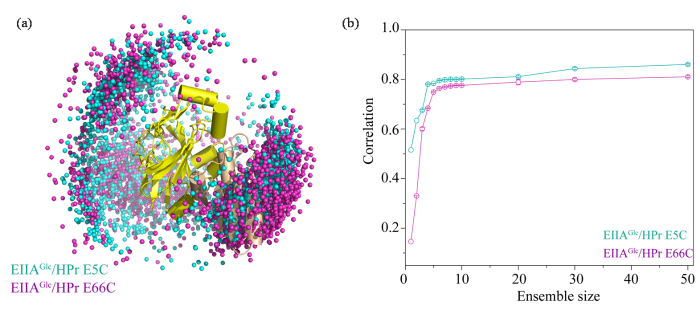
图3
(a) EIIAGlc/HPr E5C和EIIAGlc/HPr E66C复合物系综结构中HPr的空间分布;(b) PRE实验值与理论值的相关性(Correlation)随系综大小(Ensemble size)变化图
Fig. 3
(a) The spatial distributions of HPr in EIIAGlc/HPr E5C, EIIAGlc/HPr E66C complex ensemble structures; (b) The plot of ensemble size against the correlation between the experimental and the theoretical PRE value of the calculated ensemble
其中,
将两种复合物系综结构的PRE理论计算值与实验值的相关性[Correlation,图3(b)]进行对比发现,整个系综结构体系所包含的结构数量(Ensemble size)越多,PRE理论计算值与实验值的吻合性越好,在系综结构总数达到30组以上时,理论计算值与实验值的相关性保持在0.8左右,且不再发生明显改变.为避免对PRE实验数据的过拟合,我们选择包含30个构象的系综结构体系对其在溶液体系中的动态结构进行研究.
图4为EIIAGlc/HPr复合物在溶液体系中的动态分布,结果表明在EIIAGlc/HPr形成遭遇性复合物的过程中,HPr分布在EIIAGlc的特异性结合位点周围,其空间位置可大致划分为3个团簇.其中,团簇I位于EIIAGlc与HPr的特异性界面上方,相较于其他团簇更靠近于特异性结合界面,簇中的HPr与EIIAGlc的空间取向也与特异性复合物相似.而团簇II和团簇III中HPr的位置离EIIAGlc特异性活性位点相对较远,需在溶剂空间环境中经过旋转后才能与EIIAGlc的特异性位点对齐并发生蛋白质结合.遭遇复合物系综结构中各团簇空间位置的差异导致单个HPr蛋白分子与单个EIIAGlc蛋白分子间距离分布的不同,这进一步解释了图2中PRE实验值与理论计算值之间的差异.EIIAGlc/HPr团簇I靠近EIIAGlc的80~85残基,团簇II靠近EIIAGlc的110~120残基,团簇III靠近EIIAGlc的140~150残基,与EIIAGlc PRE实验值与理论计算值之间存在差异的区域大致吻合[图2(f)].
图4
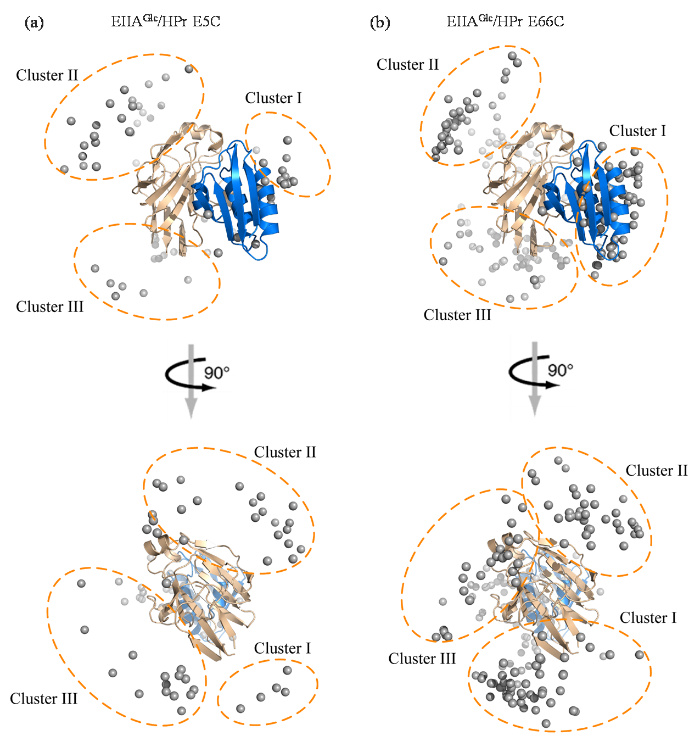
图4
EIIAGlc/HPr E5C (a)和EIIAGlc/HPr E66C (b)在缓冲液中的系综计算结构.其中包括EIIAGlc(黄色)和HPr(蓝色)的特异性复合物,EIIAGlc/HPr E5C和EIIAGlc/HPr E66C复合物中的HPr分子的质心以灰色圆圈表示.根据HPr在EIIAGlc表面的位置可将其分布方向分为三个簇,用虚线圆圈表示
Fig. 4
Calculated ensemble structures of EIIAGlc/HPr E5C (a) and EIIAGlc/HPr E66C (b) complexes in dilute buffer. The specific complex of EIIAGlc (yellow) and HPr (blue) are shown as cartoon, and the mass center of HPr molecules of the calculated EIIAGlc/HPr E5C and EIIAGlc/HPr E66C complexes are shown as gray dots. According to the location on the EIIAGlc surface, HPr molecules are divided into three clusters marked using dashed cycles
2.3 MD模拟与PRE实验结果的交叉验证
为进一步验证PRE的实验结果,我们对EIIAGlc/HPr的三种遭遇复合物结构进行了MD模拟.为了保证模拟采样的充分性,我们分别针对不同的初始结构独立模拟了三条不同的轨道(不同的随机数作为起始),从模拟的结果来看,虽然不同的模拟轨道之间有涨落,但是总体的趋势是比较一致的. 三条轨道总体来说都比较收敛,没有出现RMSD值或者距离一直在增大或一直在变化的情况. 由RMSD结果[图5(a)~(c)]可知:因为Cluster I蛋白质空间位置分布与特异性复合物相似,所以在Cluster I区域形成的遭遇复合物结构基本保持稳定;由于Cluster II与Cluster III离EIIAGlc/HPr特异性相互作用界面较远,在此区域形成的遭遇复合物的RMSD均出现较大涨落,说明在计算过程中它们偏离了模拟的初始结构.然而,在Cluster II与Cluster III模拟结果中,EIIAGlc和HPr之间的距离并没有随着模拟时间一直增大[图5(d)~(f)],说明在模拟时间内,这两种遭遇复合物并没有发生蛋白质解离,我们猜测可能是HPr绕着EIIAGlc进行旋转.模拟结构图[图5(g)~(i)]表示,HPr从起始位置(模拟时间0 ns)开始逐渐向特异性结合界面方向运动(模拟时间20 ns,60 ns).这证实了我们之前的猜想,即EIIAGlc/HPr遭遇复合物从起始位置开始,绕着EIIAGlc逐渐向EIIAGlc/HPr特异性复合物结合界面旋转,最终形成特异性复合物.目前的MD模拟描述了部分遭遇复合物动态过程,在Cluster II与Cluster III类遭遇复合物体系中,HPr由遭遇复合物起始位置开始,逐渐向特异性相互作用界面移动.
图5

图5
(a)~(c)三种遭遇复合物RMSD值随时间的变化;(d)~(f)三种遭遇复合物EIIAGlc与HPr质心间距离随时间的变化;(g)~(i)三种遭遇复合物的分子动力学模拟60 ns内结构变化示意图,其中Ec代表遭遇复合物(Encounter Complex,EC),Sc代表特异性复合物(Specific Complex,Sc)
Fig. 5
(a)~(c) The RMSD plots of three encounter complexes in three parallel trajectories; (d)~(f) The distance plots of three encounter complexes in three parallel trajectories; (g)~(i) The structure change schemes of three different encounter complexes during 60 ns under MD simulations, Ec represents encounter complex (Ec), Sc represents specific complex (Sc)
3 结论
本研究通过PRE技术及MD模拟对EIIAGlc/HPr遭遇复合物动态结构进行解析,PRE实验表明,单一的特异性复合体的构象并不能符合PRE的测量曲线,这提示EIIAGlc/HPr在溶液中存在多种构象状态,这些不同的构象状态应该是蛋白质的遭遇复合物. 通过分子对接采样,以及对PRE实验数据的拟合,我们可以获得遭遇复合物的分布位置,并通过聚类分析能把遭遇复合物分为3类.由MD模拟结果我们发现,虽然HPr远离特异性结合界面位置形成的遭遇复合物不稳定,但是复合物并未发生解离,故HPr很可能首先与EIIAGlc形成遭遇复合物,之后绕着EIIAGlc旋转到达特异性结合界面,形成特异性复合物.此分析方法不仅适用于研究蛋白质的相互作用机理,还有望应用于揭示蛋白质在生物体内的动力学行为.
致谢
感谢国家自然科学基金面上项目(31971155)的支持.
利益冲突
无
参考文献
Visualization of transient encounter complexes in protein-protein association
[J].DOI:10.1038/nature05201 [本文引用: 2]
Detecting transient intermediates in macromolecular binding by paramagnetic NMR
[J].DOI:10.1038/nature04673 [本文引用: 1]
Visualizing an ultra-weak protein-protein interaction in phosphorylation signaling
[J].DOI:10.1002/anie.201405976 URL [本文引用: 1]
Biomolecular diffusional association
[J].DOI:10.1016/S0959-440X(02)00311-1 URL [本文引用: 1]
Fundamental aspects of protein-protein association kinetics
[J].DOI:10.1021/cr800373w PMID:19196002 [本文引用: 1]
The courtship of proteins: understanding the encounter complex
[J].
DOI:10.1016/j.febslet.2009.02.046
PMID:19275897
[本文引用: 1]

The formation of protein complexes involves an encounter complex, in which proteins show few specific interactions and assume many orientations. Recent kinetic and structural studies have shed light on this elusive state. It is generally dominated by electrostatic interactions, although hydrophobic interactions can play a role. During the encounter phase the proteins remain largely solvated. In extreme cases, the proteins only form an encounter complex, and in many other complexes, the encounter state constitutes a significant amount (5% or more), indicating that the energy difference between encounter and productive complexes is small. Thus, the encounter complex represents an essential part of protein complexes.
The transient complex of cytochrome c and cytochrome c peroxidase: Insights into the encounter complex from multifrequency EPR and NMR spectroscopy
[J].
DOI:10.1002/cphc.201901160
PMID:32301564
[本文引用: 1]
We present a novel approach to study transient protein-protein complexes with standard, 9 GHz, and high-field, 95 GHz, electron paramagnetic resonance (EPR) and paramagnetic NMR at ambient temperatures and in solution. We apply it to the complex of yeast mitochondrial iso-1-cytochrome c (Cc) with cytochrome c peroxidase (CcP) with the spin label [1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)-methanethiosulfonate] attached at position 81 of Cc (SL-Cc). A dissociation constant K of 20±4×10 M (EPR and NMR) and an equal amount of stereo-specific and encounter complex (NMR) are found. The EPR spectrum of the fully bound complex reveals that the encounter complex has a significant population (60 %) that shares important features, such as the Cc-interaction surface, with the stereo-specific complex.© 2020 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA.
Brownian dynamics of cytochrome c and cytochrome c peroxidase association
[J].Brownian dynamics computer simulations of the diffusional association of electron transport proteins cytochrome c (cyt c) and cytochrome c peroxidase (cyt c per) were performed. A highly detailed and realistic model of the protein structures and their electrostatic interactions was used that was based on an atomic-level spatial description. Several structural features played a role in enhancing and optimizing the electron transfer efficiency of this reaction. Favorable electrostatic interactions facilitated long-lived nonspecific encounters between the proteins that allowed the severe orientational criteria for reaction to be overcome by rotational diffusion during encounters. Thus a "reduction-in-dimensionality" effect operated. The proteins achieved plausible electron transfer orientations in a multitude of electrostatically stable encounter complexes, rather than in a single dominant complex.
Rapid, electrostatically assisted association of proteins
[J].The rapid association of barnase and its intracellular inhibitor barstar has been analysed from the effects of mutagenesis and electrostatic screening. A basal association rate constant of 10(5) M(-1) s(-1) is increased to over 5 x 10(9) M(-1) s(-1) by electrostatic forces. The association between the oppositely charged proteins proceeds through the rate-determining formation of an early, weakly specific complex, which is dominated by long-range electrostatic interactions, followed by precise docking to form the high affinity complex. This mode of binding is likely to be used widely in nature to increase association rate constants between molecules and its principles may be used for protein design.
Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar
[J].The electrostatic enhancement of the association rate of barnase and barstar is calculated using a transition-state theory like expression and atomic-detail modeling of the protein molecules. This expression predicts that the rate enhancement is simply the average Boltzmann factor in the region of configurational space where association occurs instantaneously in the diffusion-controlled limit. Based on experimental evidence, this "transition state" is defined by configurations in which, relative to the stereospecifically bound complex, the two proteins are shifted apart by approximately 8 A (so a layer of water can be accommodated in the interface) and the two binding surfaces are rotated away by 0 degrees to 3 degrees. The values of the average Boltzmann factor, calculated by solving the Poisson-Boltzmann equation, for the wild-type complex and 16 complexes with single mutations are found to correlate well with experimental results for the electrostatic rate enhancement. The predicted rate enhancement is found to be somewhat insensitive to the precise definition of the transition state, due to the long-range nature of electrostatic interactions. The experimental ionic strength dependence of the rate enhancement is also reasonably reproduced.Copyright 1998 Academic Press Limited.
Rational design of faster associating and tighter binding protein complexes
[J].A protein design strategy was developed to specifically enhance the rate of association (k(on)) between a pair of proteins without affecting the rate of dissociation (k(off)). The method is based on increasing the electrostatic attraction between the proteins by incorporating charged residues in the vicinity of the binding interface. The contribution of mutations towards the rate of association was calculated using a newly developed computer algorithm, which predicted accurately the rate of association of mutant protein complexes relative to the wild type. Using this design strategy, the rate of association and the affinity between TEM1 beta-lactamase and its protein inhibitor BLIP was enhanced 250-fold, while the dissociation rate constant was unchanged. The results emphasize that long range electrostatic forces specifically alter k(on), but do not effect k(off). The design strategy presented here is applicable for increasing rates of association and affinities of protein complexes in general.
Enhancement of association rates by nonspecific binding to DNA and cell membranes
[J].DOI:10.1103/PhysRevLett.93.178101 URL [本文引用: 1]
On the dynamic nature of the transition state for protein-protein association as determined by double-mutant cycle analysis and simulation
[J].The process of protein-protein association starts with their random collision, which may develop into an encounter complex followed by a transition state and final complex formation. Here we aim to experimentally characterize the nature of the transition state of protein-protein association for three different protein-protein interactions; Barnase-Barstar, TEM1-BLIP and IFNalpha2-IFNAR2, and use the data to model the transition state structures. To model the transition state, we determined inter-protein distance-constraints of the activated complex by using double mutant cycles (DMC) assuming that interacting residues are spatially close. Significant DeltaDeltaG(double dagger)(int) values were obtained only between residues on Barnase and Barstar. However, introducing specific mutations that optimize the charge complementarity between BLIP and TEM1 resulted in the introduction of significant DeltaDeltaG(double dagger)(int) values also between residues of these two proteins. While electrostatic interactions make major contributions towards stabilizing the transition state, we show two examples where steric hindrance exerts an effect on the transition state as well. To model the transition-state structures from the experimental DeltaDeltaG(double dagger)(int) values, we introduced a method for structure perturbation, searching for those inter-protein orientations that best support the experimental DeltaDeltaG(double dagger)(int) values. Two types of transition states were found, specific as observed for Barnase-Barstar and the electrostatically optimized TEM1-BLIP mutants, and diffusive as shown for wild-type TEM1-BLIP and IFNalpha2-IFNAR2. The specific transition states are characterized by defined inter-protein orientations, which cannot be modeled for the diffusive transition states. Mutations introduced through rational design can change the transition state from diffusive to specific. Together, these data provide a structural view of the mechanism allowing rates of association to differ by five orders of magnitude between different protein complexes.
Asymmetric catalytic hydrogenation. Design of new Ru catalysts and chiral ligands: from laboratory to industrial applications
[J].DOI:10.1021/ar020152u URL [本文引用: 1]
Myoglobin and cytochrome b5: a nuclear magnetic resonance study of a highly dynamic protein complex
[J].The transient complex of bovine myoglobin and cytochrome b(5) has been investigated using a combination of NMR chemical shift mapping, (15)N relaxation data, and protein docking simulations. Chemical shift perturbations observed for cytochrome b(5) amide resonances upon complex formation with either metmyoglobin (Fe(III)) or carbon monoxide-bound myoglobin (Fe(II)) are more than 10-fold smaller than in other transient redox protein complexes. From (15)N relaxation experiments, an increase in the overall correlation time of cytochrome b(5) in the presence of myoglobin is observed, confirming that complex formation is occurring. The chemical shift perturbations of proton and nitrogen amide nuclei as well as heme protons of cytochrome b(5) titrate with increasing myoglobin concentrations, also demonstrating the formation of a weak complex with a K(a) in the inverse millimolar range. The perturbed residues map over a wide surface area of cytochrome b(5), with patches of residues located around the exposed heme 6-propionate as well as at the back of the protein. The nature of the affected residues is mostly negatively charged contrary to perturbed residues in other transient complexes, which are mainly hydrophobic or polar. Protein docking simulations using the NMR data as constraints show several docking geometries both close to and far away from the exposed heme propionates of myoglobin. Overall, the data support the emerging view that this complex consists of a dynamic ensemble of orientations in which each protein constantly diffuses over the surface of the other. The characteristic NMR features may serve as a structural tool for the identification of such dynamic complexes.
Complex of plastocyanin and cytochrome c characterized by NMR chemical shift analysis
[J].The complexes of horse ferrous and ferric cytochrome c with Cd-substituted pea plastocyanin have been characterized by nuclear magnetic resonance, in order to determine the binding sites and to study the effects of complex formation. Reproducible, small chemical shift changes (0.005-0.05 ppm) were observed for protons in both proteins upon formation of a 1:1 complex. The chemical shift changes depended on the ratio of free to bound protein, with a binding constant of 1.0 +/- 0.5 x 10(5) M(-1), indicating that they were caused by complex formation and that free and bound proteins were in fast exchange. Two-dimensional spectra of the complex of ferrocytochrome c and plastocyanin were screened systematically for chemical shift changes. For about 760 protons, or 70% of the assigned protons in the two proteins, the chemical shift in the complex could be established. In plastocyanin and cytochrome c 14% and 17% of the protons, respectively, showed a significant chemical shift change. These protons form two groups. The first consists of a limited number of surface-exposed side-chain protons. These map on the so-called east side of plastocyanin and the front side of cytochrome c. This group of chemical shift changes is interpreted as representing direct effects of binding, and the respective surfaces thus represent the binding sites. The second group includes backbone amide protons and a few aliphatic and aromatic protons in the hydrophobic core of each protein. The chemical shift changes of this group are interpreted as secondary, i.e., caused by very small structural changes which are transmitted deep into the core of the protein. Ferric cytochrome c caused the same chemical shift effects in plastocyanin as the ferrous form; no intermolecular paramagnetic effects were observed. The small size of the chemical shifts and the absence of intermolecular paramagnetic shifts and NOEs suggest that the complex consists of a dynamic ensemble of structures which are in fast exchange, rather than a single static complex. This study shows that small, reproducible chemical shifts can be used effectively to characterize protein complexes in detail.
Transient protein interactions studied by NMR spectroscopy: the case of cytochrome C and adrenodoxin
[J].The interaction between yeast iso-1-cytochrome c (C102T) and two forms of bovine adrenodoxin, the wild type and a truncated form comprising residues 4-108, has been investigated using a combination of one- and two-dimensional heteronuclear NMR spectroscopy. Chemical shift perturbations and line broadening of amide resonances in the [(15)N,(1)H]HSQC spectrum for both (15)N-labeled cytochrome c and adrenodoxin in the presence of the unlabeled partner protein indicate the formation of a transient complex, with a K(a) of (4 +/- 1) x 10(4) M(-)(1) and a lifetime of <3 ms. The perturbed residues map over a large surface area for both proteins. For cytochrome c, the dominating effects are located around the exposed heme edge but with other areas also affected upon formation of the complex. In the case of adrenodoxin, effects are seen in both the recognition and core domains, with the largest perturbations in the recognition domain. These results indicate that the complex has a dynamic nature, with delocalized binding of cytochrome c on adrenodoxin. A comparison with other transient complexes of redox proteins places this complex between well-defined complexes such as the cytochrome c-cytochrome c peroxidase complex and entirely dynamic complexes such as the cytochrome b(5)-myoglobin complex.
Dynamics in electron transfer protein complexes
[J].
DOI:10.1111/j.1742-4658.2011.08062.x
PMID:21352493
[本文引用: 1]
Electron transfer proteins transport electrons safely between large redox enzymes. The complexes formed by these proteins are among the most transient. The biological function requires, on the one hand, sufficient specificity of the interaction to allow for rapid and selective electron transfer, and, on the other hand, a fast turnover of the complex. Recent progress in the characterization of the nature of these complexes has demonstrated that the encounter state plays an important role. This state of initial binding is dominated by electrostatic interactions, and consists of an ensemble of orientations. Paramagnetic relaxation enhancement NMR and chemical shift perturbation analysis provide ways for the experimental characterisation of the encounter state. Several studies that have used these techniques have shown that the surface area sample in the encounter state can be limited to the immediate environment of the final, specific complex. The encounter complex can represent a large fraction and, in some small complexes, no specific binding is detected at all. It can be concluded that, in electron transfer protein complexes, a fine balance is sought between the low-specificity encounter state and the high-specificity productive complex to meet the opposing requirements of rapid electron transfer and a high turnover rate.© 2011 The Authors Journal compilation © 2011 FEBS.
Mechanisms of chaperones as active assistant/protector for proteins: Insights from NMR studies
[J].DOI:10.1002/cjoc.v38.4 URL [本文引用: 1]
NMR evidence for slow collective motions in cyanometmyoglobin
[J].Residual dipolar couplings observed in NMR spectra at very high magnetic fields have been measured to a high degree of accuracy for the paramagnetic protein cyanometmyoglobin. Deviations of these measurements from predictions based on available crystallographic and solution structures are largely systematic and well correlated within a given helix of this highly alpha-helical protein. These observations can be explained by invoking collective motion and small displacements of representative helices from their reported average positions in the solid state. Thus, the measurements appear to be capable of providing important insights into slower, collective protein motions, which are likely to be important for function, and which have been difficult to study using established experimental techniques.
NMR assignments and characterization of the DNA-binding domain of Arabidopsis transcription factor WRKY11
[J].
New approaches for computing ligand-receptor binding kinetics
[J].DOI:10.1016/j.sbi.2017.10.001 URL [本文引用: 1]
Kinetics of ligand binding through advanced computational approaches: A review
[J].
DOI:10.2174/1568026617666170414142908
PMID:28413946
[本文引用: 1]
Ligand residence times and binding rates have been found to be useful quantities to consider during drug design. The underlying structural and dynamic determinants of these kinetic quantities are difficult to discern. Driven by developments in computational hardware and simulation methodologies, molecular dynamics (MD) studies on full binding and unbinding pathways have emerged recently, showing these structural and dynamic determinants in atomic detail. However, the long timescales related to drug binding and release are still prohibitive to conventional MD simulation. Here we discuss a suite of enhanced sampling methods that have been applied to the study of full ligand binding or unbinding pathways, and reveal the kinetics of drug binding and/or release. We divide these sampling methods into three families (trajectory parallelization, metadynamics, and temperature- based methods), and discuss recent applications of each, as well as their basic theoretical underpinnings including how kinetic information is extracted. We then present an outlook for how the field could evolve, and how the rich variety of sampling methods discussed here can be leveraged in the future for computationally driven drug design.Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.org.
Role of molecular dynamics and related methods in drug discovery
[J].
DOI:10.1021/acs.jmedchem.5b01684
PMID:26807648
[本文引用: 1]
Molecular dynamics (MD) and related methods are close to becoming routine computational tools for drug discovery. Their main advantage is in explicitly treating structural flexibility and entropic effects. This allows a more accurate estimate of the thermodynamics and kinetics associated with drug-target recognition and binding, as better algorithms and hardware architectures increase their use. Here, we review the theoretical background of MD and enhanced sampling methods, focusing on free-energy perturbation, metadynamics, steered MD, and other methods most consistently used to study drug-target binding. We discuss unbiased MD simulations that nowadays allow the observation of unsupervised ligand-target binding, assessing how these approaches help optimizing target affinity and drug residence time toward improved drug efficacy. Further issues discussed include allosteric modulation and the role of water molecules in ligand binding and optimization. We conclude by calling for more prospective studies to attest to these methods' utility in discovering novel drug candidates.
Protein-ligand (un)binding kinetics as a new paradigm for drug discovery at the crossroad between experiments and modelling
[J].
DOI:10.1039/c6md00581k
PMID:30108770
[本文引用: 1]
In the last three decades, protein and nucleic acid structure determination and comprehension of the mechanisms, leading to their physiological and pathological functions, have become a cornerstone of biomedical sciences. A deep understanding of the principles governing the fates of cells and tissue at the molecular level has been gained over the years, offering a solid basis for the rational design of drugs aimed at the pharmacological treatment of numerous diseases. Historically, affinity indicators ( and IC/EC) have been assumed to be valid indicators of the efficacy of a drug. However, recent studies pointed out that the kinetics of the drug-receptor binding process could be as important or even more important than affinity in determining the drug efficacy. This eventually led to a growing interest in the characterisation and prediction of the rate constants of protein-ligand association and dissociation. For instance, a drug with a longer residence time can kinetically select a given receptor over another, even if the affinity for both receptors is comparable, thus increasing its therapeutic index. Therefore, understanding the molecular features underlying binding and unbinding processes is of central interest towards the rational control of drug binding kinetics. In this review, we report the theoretical framework behind protein-ligand association and highlight the latest advances in the experimental and computational approaches exploited to investigate the binding kinetics.
How does a drug molecule find its target binding site?
[J].
DOI:10.1021/ja202726y
PMID:21545110
[本文引用: 1]
Although the thermodynamic principles that control the binding of drug molecules to their protein targets are well understood, detailed experimental characterization of the process by which such binding occurs has proven challenging. We conducted relatively long, unguided molecular dynamics simulations in which a ligand (the cancer drug dasatinib or the kinase inhibitor PP1) was initially placed at a random location within a box that also contained a protein (Src kinase) to which that ligand was known to bind. In several of these simulations, the ligand correctly identified its target binding site, forming a complex virtually identical to the crystallographically determined bound structure. The simulated trajectories provide a continuous, atomic-level view of the entire binding process, revealing persistent and noteworthy intermediate conformations and shedding light on the role of water molecules. The technique we employed, which does not assume any prior knowledge of the binding site's location, may prove particularly useful in the development of allosteric inhibitors that target previously undiscovered binding sites.
Pathway and mechanism of drug binding to G-protein-coupled receptors
[J].
DOI:10.1073/pnas.1104614108
URL
[本文引用: 1]
\n How drugs bind to their receptors—from initial association, through drug entry into the binding pocket, to adoption of the final bound conformation, or “pose”—has remained unknown, even for G-protein-coupled receptor modulators, which constitute one-third of all marketed drugs. We captured this pharmaceutically critical process in atomic detail using the first unbiased molecular dynamics simulations in which drug molecules spontaneously associate with G-protein-coupled receptors to achieve final poses matching those determined crystallographically. We found that several beta blockers and a beta agonist all traverse the same well-defined, dominant pathway as they bind to the\n β\n 1\n - and\n β\n 2\n -adrenergic receptors, initially making contact with a vestibule on each receptor’s extracellular surface. Surprisingly, association with this vestibule, at a distance of 15 Å from the binding pocket, often presents the largest energetic barrier to binding, despite the fact that subsequent entry into the binding pocket requires the receptor to deform and the drug to squeeze through a narrow passage. The early barrier appears to reflect the substantial dehydration that takes place as the drug associates with the vestibule. Our atomic-level description of the binding process suggests opportunities for allosteric modulation and provides a structural foundation for future optimization of drug–receptor binding and unbinding rates.\n
Dissociation process of a MDM2/p53 complex investigated by parallel cascade selection molecular dynamics and the markov state model
[J].DOI:10.1021/acs.jpcb.8b10309 URL [本文引用: 1]
Mapping the ligand binding landscape
[J].
DOI:S0006-3495(18)31102-0
PMID:30327139
[本文引用: 1]
The interaction between a ligand and a protein involves a multitude of conformational states. To achieve a particular deeply bound pose, the ligand must search across a rough free-energy landscape with many metastable minima. Creating maps of the ligand binding landscape is a great challenge, as binding and release events typically occur on timescales that are beyond the reach of molecular simulation. The WExplore enhanced sampling method is well suited to build these maps because it is designed to broadly explore free-energy landscapes and is capable of simulating ligand release pathways that occur on timescales as long as minutes. WExplore also uses only unbiased trajectory segments, allowing for the construction of Markov state models (MSMs) and conformation space networks that combine the results of multiple simulations. Here, we use WExplore to study two bromodomain-inhibitor systems using multiple docked starting poses (Brd4-MS436 and Baz2B-ICR7) and synthesize our results using a series of MSMs using time-lagged independent component analysis. Ranking the starting poses by exit rate agrees with the crystal structure pose in both cases. We also predict the most stable pose using the equilibrium populations from the MSM but find that the prediction is not robust as a function of MSM parameters. The simulated trajectories are synthesized into network models that visualize the entire binding landscape for each system, and we examine transition paths between deeply bound stable states. We find that, on average, transitions between deeply bound states convert through the unbound state 81% of the time, implying a trial-and-error approach to ligand binding. We conclude with a discussion of the implications of this result for both kinetics-based drug discovery and virtual screening pipelines that incorporate molecular dynamics.Copyright © 2018 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models
[J].
DOI:10.1038/ncomms8653
PMID:26134632
[本文引用: 1]
Plattner, Nuria; Noe, Frank Free Univ Berlin, Dept Math Comp Sci & Bioinformat, D-14195 Berlin, Germany.
A role for both conformational selection and induced fit in ligand binding by the LAO protein
[J].DOI:10.1371/journal.pcbi.1002054 URL [本文引用: 1]
The phosphoenolpyruvate:sugar phosphotransferase system in gram-positive bacteria: properties, mechanism, and regulation
[J].This review consists of three major sections. The first and largest section reviews the protein constituents and known properties of the phosphotransferase systems present in well-studied Gram-positive bacteria. These bacteria include species of the following genera: (1) Staphylococcus, (2) Streptococcus, (3) Bacillus, (4) Lactobacillus, (5) Clostridium, (6) Arthrobacter, and (7) Brochothrix. The properties of the different systems are compared. The second major section deals with the regulation of carbohydrate uptake. There are four parts: (1) inhibition by intracellular sugar phosphates in Staphylococcus aureus, (2) PTS-mediated regulation of glycerol uptake in Bacillus subtilis, (3) competition for phospho-HPr in Streptococcus mutans, and (4) the possible involvement of protein kinases in the regulation of sugar uptake via the phosphotransferase system. The third section deals with the phenomenon of inducer expulsion. The first part is concerned with the physiological characterization of the phenomenon; then the consequences of unregulated uptake and expulsion, a futile cycle of energy expenditure, are considered. Finally, the biochemistry of the protein kinase and the protein phosphate phosphatase system, which appears to regulate sugar transport via the phosphotransferase system, is defined. The review, therefore, concentrates on the phosphotransferase system, its functions in carbohydrate transport and phosphorylation, the mechanisms of its regulation, and the mechanism by which it participates in the regulation of other physiological processes in the bacterial cell.
Unraveling a bacterial hexose transport pathway
[J].DOI:10.1016/0959-440X(94)90262-3 URL [本文引用: 1]
Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIA(glucose) of the Escherichia coli phosphoenolpyruvate: sugar phosphotransferase system
[J].DOI:10.1093/emboj/19.21.5635 URL [本文引用: 1]
Visualizing the ensemble structures of protein complexes using chemical cross-linking coupled with mass spectrometry
[J].DOI:10.1007/s41048-015-0015-y URL [本文引用: 1]
Mechanistic details of a protein-protein association pathway revealed by paramagnetic relaxation enhancement titration measurements
[J].
DOI:10.1073/pnas.0909370107
URL
[本文引用: 1]
\n Protein–protein association generally proceeds via the intermediary of a transient, lowly populated, encounter complex ensemble. The mechanism whereby the interacting molecules in this ensemble locate their final stereospecific structure is poorly understood. Further, a fundamental question is whether the encounter complex ensemble is an effectively homogeneous population of nonspecific complexes or whether it comprises a set of distinct structural and thermodynamic states. Here we use intermolecular paramagnetic relaxation enhancement (PRE), a technique that is exquisitely sensitive to lowly populated states in the fast exchange regime, to characterize the mechanistic details of the transient encounter complex interactions between the N-terminal domain of Enzyme I (EIN) and the histidine-containing phosphocarrier protein (HPr), two major bacterial signaling proteins. Experiments were conducted at an ionic strength of 150 mM NaCl to eliminate any spurious nonspecific associations not relevant under physiological conditions. By monitoring the dependence of the intermolecular transverse PRE (Γ\n 2\n ) rates measured on\n 15\n N-labeled EIN on the concentration of paramagnetically labeled HPr, two distinct types of encounter complex configurations along the association pathway are identified and dissected. The first class, which is in equilibrium with and sterically occluded by the specific complex, probably involves rigid body rotations and small translations near or at the active site. In contrast, the second class of encounter complex configurations can coexist with the specific complex to form a ternary complex ensemble, which may help EIN compete with other HPr binding partners in vivo by increasing the effective local concentration of HPr even when the active site of EIN is occupied.\n
Modeling protein excited-state structures from “over-length” chemical cross-links
[J].DOI:10.1074/jbc.M116.761841 URL [本文引用: 1]
Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIA(glucose) of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system
[J].The solution structure of the second protein-protein complex of the Escherichia coli phosphoenolpyruvate: sugar phosphotransferase system, that between histidine-containing phosphocarrier protein (HPr) and glucose-specific enzyme IIA(Glucose) (IIA(Glc)), has been determined by NMR spectroscopy, including the use of dipolar couplings to provide long-range orientational information and newly developed rigid body minimization and constrained/restrained simulated annealing methods. A protruding convex surface on HPr interacts with a complementary concave depression on IIA(Glc). Both binding surfaces comprise a central hydrophobic core region surrounded by a ring of polar and charged residues, positive for HPr and negative for IIA(Glc). Formation of the unphosphorylated complex, as well as the phosphorylated transition state, involves little or no change in the protein backbones, but there are conformational rearrangements of the interfacial side chains. Both HPr and IIA(Glc) recognize a variety of structurally diverse proteins. Comparisons with the structures of the enzyme I-HPr and IIA(Glc)-glycerol kinase complexes reveal how similar binding surfaces can be formed with underlying backbone scaffolds that are structurally dissimilar and highlight the role of redundancy and side chain conformational plasticity.
QuikChange shuffling: a convenient and robust method for site-directed mutagenesis and random recombination of homologous genes
[J].DOI:10.1016/j.nbt.2011.03.001 URL [本文引用: 1]
The Xplor-NIH NMR molecular structure determination package
[J].We announce the availability of the Xplor-NIH software package for NMR biomolecular structure determination. This package consists of the pre-existing XPLOR program, along with many NMR-specific extensions developed at the NIH. In addition to many features which have been developed over the last 20 years, the Xplor-NIH package contains an interface with a new programmatic framework written in C++. This interface currently supports the general purpose scripting languages Python and TCL, enabling rapid development of new tools, such as new potential energy terms and new optimization methods. Support for these scripting languages also facilitates interaction with existing external programs for structure analysis, structure manipulation, visualization, and spectral analysis.
ZDOCK: an initial-stage protein-docking algorithm
[J].DOI:10.1002/(ISSN)1097-0134 URL [本文引用: 1]
Ensemble approach for NMR structure refinement against (1)H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule
[J].Paramagnetic relaxation enhancement (PRE) measurements on (1)H nuclei have the potential to play an important role in NMR structure determination of macromolecules by providing unique long-range (10-35 A) distance information. Recent methodological advances for covalently attaching paramagnetic groups at specific sites on both proteins and nucleic acids have permitted the application of the PRE to various biological macromolecules. However, because artificially introduced paramagnetic groups are exposed to solvent and linked to the macromolecule by several freely rotatable bonds, they are intrinsically flexible. This renders conventional back-calculation of the (1)H-PRE using a single-point representation inaccurate, thereby severely limiting the utility of the (1)H-PRE as a tool for structure refinement. To circumvent these limitations, we have developed a theoretical framework and computational strategy with which to accurately back-calculate (1)H-PREs arising from flexible paramagnetic groups attached to macromolecules. In this scheme, the (1)H-PRE is calculated using a modified Solomon-Bloembergen equation incorporating a "model-free" formalism, based on a multiple-structure representation of the paramagnetic group in simulated annealing calculations. The ensemble approach for (1)H-PRE back-calculation was examined using several SRY/DNA complexes incorporating dT-EDTA-Mn(2+) at three distinct sites in the DNA, permitting a large data set comprising 435 experimental backbone and side-chain (1)H-PREs to be obtained in a straightforward manner from 2D through-bond correlation experiments. Calculations employing complete cross-validation demonstrate that the ensemble representation provides a means to accurately utilize backbone and side-chain (1)H-PRE data arising from a flexible paramagnetic group in structure refinement. The results of (1)H-PRE based refinement, in conjunction with previously obtained NMR restraints, indicate that significant gains in accuracy can be readily obtained. This is particularly significant in the case of macromolecular complexes where intermolecular translational restraints derived from nuclear Overhauser enhancement data may be limited.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}