


引言
药物杂质是指药物制剂中存在的无治疗作用或者影响药物的稳定性、疗效,甚至对人体健康有害的物质[1-2]. 药物杂质来源多样,在药品生产、包装、流通及存储等多个环节都可能产生,难以预测,这使得对其的定性分析成为一种挑战. 只有确定药物杂质化学结构,才可能了解其形成机制及毒理学性质,进而优化制剂生产工艺,降低杂质含量到可接受的水平,才能保证药物的有效性和安全性. 现代分析化学技术的进步,为药物杂质分析提供了便利,其中现代仪器分析技术在药物杂质分析中发挥着重要作用. 药物杂质鉴定,通常以色谱分离方法制备出足够量的药物杂质,然后进行核磁共振(NMR)与质谱(MS)等谱学测试,最后鉴定出化学结构.随着电喷雾离子源(ESI)技术的发展与应用,液相色谱-质谱(LC-MS)联用技术具有高效分离能力、高灵敏与快速定性能力而被用于药物杂质快速定性分析[3⇓⇓⇓-7]. 而电喷雾离子源存在离子抑制、离子化效率等问题,以及质谱定性能力不及NMR技术,往往不能单独用于结构鉴定. NMR技术具有卓越的有机化合物结构解析能力,因此液相色谱-核磁共振(LC-NMR)联用技术也被尝试用于药物杂质分析[8,9]. 早期NMR技术灵敏度较低,以及药物杂质含量水平较低,使得LC-NMR联用技术难以用于微量定性分析. 因此,药物杂质分析往往运用多种分析化学技术进行综合解析,例如LC-MS技术与制备级液相色谱,制备出的杂质后进行NMR测试,最终完成药物杂质化学结构解析.
本世纪初发展出的液相色谱-二极管阵列检测器-固相萃取-核磁共振/质谱(LC-DAD-SPE-NMR/MS)联用技术[10],集成了LC、SPE、NMR与MS等现代分析化学技术的优势,用于复杂样品体系中有机化合物的定性分析. 该技术中SPE技术可对高效液相色谱仪洗脱出的色谱峰进行多次自动化在线固相萃取以浓缩样品,然后以氘代试剂将萃取物从固相萃取柱上洗脱并将溶液转移至NMR谱仪的流动探头中进行NMR测试. 该联用技术推荐使用高灵敏氦气超低温探头的NMR谱仪,与常温探头相比,理论上超低温探头可将信噪比提高到4倍,因此使得样品需求量减少至常温探头的1/4,更适合于微量样品的检测,并使得高质量一维与二维NMR谱的采集成为可能. 联用系统中的MS可用于复杂样品体系中化学成分的分子量快速测定,为结构类型、化学结构快速判断与验证NMR解析结果提供了可能. LC-DAD-SPE-NMR/MS联用技术的推出,使得NMR检测对样品量的需求大为降低,可用于微量成分的定性分析,因而被用于复杂样品体系中化合物定性分析等应用场景[11⇓⇓⇓⇓⇓-17],该联用技术在植物化学研究[10,12,16,17]中的应用证实该技术具有强大分析能力. 此联用技术曾被用于药物杂质分析[18⇓-20],其中Harča等采用该技术鉴定了化学原料药3-溴-5-(三氟甲基)苯胺中含量在0.37%~1.33%之间的三个主要杂质的化学结构[20],表明此技术可应用于低含量药物杂质的结构鉴定.
卡巴他赛(C45H57NO14,835.93)(图1)是由赛诺菲制药公司研发的半合成紫杉烷类小分子化合物类微管抑制剂[21],2010年经美国食品药品监督管理局批准上市销售. 该药适合与泼尼松或泼尼松龙联合用于多西他赛治疗方案失败的难治转移性前列腺癌患者. 某制药公司在仿制卡巴他赛注射液时,选用了与赛诺菲公司不同化学合成途径的原料药,在加速稳定试验分析中,LC检测到一个未见文献报道[22⇓-24]的降解杂质,含量约为0.3%~0.4%. 因卡巴他赛水溶性较差,其注射液处方中添加乙醇和高比例的吐温-80等辅料以保证生产、存储和临床注射过程中卡巴他赛有稳定的溶解度. 吐温-80是由山梨醇酐单甘油酸酯和环氧乙烷共聚制得的聚合物,化学结构中失水山梨醇上4个羟基上共连接20个环氧乙烷基团而形成的一类混合物,在液相色谱分析中出现一系列色谱峰,表明失水山梨醇上不同羟基连接不同环氧乙烷基团后疏水性能有明显差异. 作为一种非离子型表面活性剂,其化学结构中没有明显的紫外生色基团,因此采用紫外检测器难以检出低浓度的吐温-80类成分. LC-MS分析中发现吐温-80与降解杂质峰一同洗脱出色谱柱,表明降解杂质与吐温-80疏水性相似,因此采用制备LC方法难以分离得到高纯度降解杂质,影响降解杂质结构解析. 本研究尝试采用LC-DAD-SPE-NMR/MS联用技术,首先通过LC-DAD-ESI-QTOF-MS模式确定降解杂质为卡巴他赛同分异构体,然后通过多次自动化在线固相萃取的方法,萃取出降解杂质色谱峰,最后进行一维与二维NMR谱测试. 萃取物溶液1H NMR谱中,吐温-80谱峰强度远高于降解杂质,掩盖降解杂质部分谱峰,本文基于NMR技术的混合物分析方法对1H-1H COSY、1H-1H NOESY、1H-13C HSQC与1H-13C HMBC等二维谱细致分析,提取出降解杂质中氢-氢、氢-碳原子间相关信息,得到氢、碳原子化学位移及连接关系,成功排除吐温-80的谱峰干扰,最终鉴定出降解杂质的化学结构. 结合文献,推测该降解杂质为卡巴他赛紫杉烷母核不饱和六元环中C-15在酸性条件下形成碳正离子,C-15及两个甲基(C-16与C-17)重排移位至C-1位而产生(图1). 此降解杂质化学结构的鉴定,为卡巴他赛注射液处方优化提供了线索.
图1
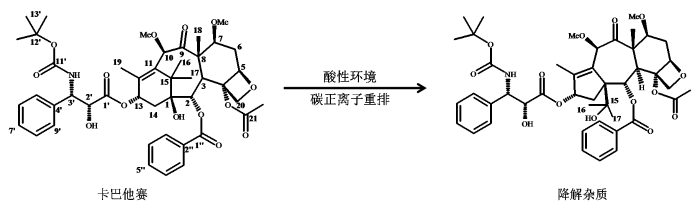
图1
卡巴他赛及其新降解杂质化学结构及其降解机制. *降解杂质中除15-17位碳原子外,其余碳原子编号与卡巴他赛相同
Fig. 1
Chemical structures of cabazitaxel and its degradation impurity and the possible formation mechanism. * The number of carbon atoms of the degradation impurity is the same as of cabazitaxel besides C-15, C-16, and C-17
1 实验部分
1.1 化学试剂与样品
色谱纯乙腈、甲醇与异丙醇为美国Tedia(Fairfield,OH,USA)产品. 分析纯甲酸为国药试剂公司产品. 氘代乙腈(99.8%)为德国Deutero GmbH(Kastellaun, Germany)产品. 超纯水为实验室Milli-Q (Milfold,MA)纯水制备系统制备.
卡巴他赛注射液(6 mL,10 mg/mL)样品为某制药公司加速稳定性试验样品,根据高效液相色谱图中卡巴他赛与未知杂质峰峰面积进行归一化法估算,降解杂质含量为0.3%~0.4%.
1.2 LC-DAD-SPE-NMR/MS联用系统
LC-DAD-SPE-NMR/MS联用系统中液相色谱仪为Agilent 1200型分析型液相色谱(Waldbronn,Germany),配备在线脱气机(G1322A)、四元泵(G1311A)、自动进样器(G1367B)和二极管阵列检测器(G1315A),以及Bruker公司BMSO柱温箱. 高分辨质谱为Bruker Daltonic公司配备ESI源的micrOTOF_Q四极杆飞行时间串联质谱仪(Bremen,Germany). 在线自动SPE仪为德国Bruker与荷兰Spark公司(Emmen,Holland)联合开发的Prospekt 2 SPE unit单元. NMR谱仪为配备5 mm 1H-13C-15N CPTCI超低温探头的Bruker Avance III 600 MHz. LC-DAD-SPE-NMR联用实验配备60 μL流通池的CryoFit插件. 联用系统所用的控制软件为Bruker公司Compass程序包,包含色谱控制软件Hystar 3.2、质谱数据采集软件micrOTOF control以及NMR数据采集软件Topspin 3.5.7.
1.3 实验过程
1.3.1 LC-DAD-ESI-QTOF-MS实验
液相色谱分离在Agilent 1200液相色谱仪上运行,色谱条件沿用该制药公司卡巴他赛注射液质量控制液相色谱分析条件:Agilent公司Zorbax SB C18反相色谱柱(250×4.6 mm,5 µm),流速1.0 mL/min,柱温35 ˚C,进样体积2 μL,检测波长230 nm. 流动相为42% A相(色谱纯乙腈)、26% B相(色谱纯甲醇)和32% C相(超纯水)组成的混合溶剂,进行等度洗脱. 洗脱液经过T形三通分流5 %洗脱液经Bruker BNMI接口后进入电喷雾离子源,而95 %洗脱液直接流入二极管阵列检测器进行紫外检测. 记录30.0 min紫外色谱与总离子流图谱. MicrOTOF-Q质谱仪离子源雾化压力0.8 Bar氮气,干燥气为8.0 L/min流速的200 ˚C氮气,离子源毛细管电压为-3 600 V,挡板电压(endplate offset)为500 V,正离子模式采集,质量范围为m/z 50~1 700. 本实验数据采集使用Hystar 3.2和micrOTOFcontrol,数据分析使用DataAnalysis 4.0.
1.3.2 LC-DAD-SPE自动在线萃取
LC-DAD-SPE自动在线萃取用于注射液中降解杂质的富集. 色谱洗脱程序与LC-DAD-ESI-QTOF-MS分析相同,注射液样品进样体积改为100 μL. 注射液中含高比例的吐温-80导致溶液粘度较大,因此设定液相色谱自动进样器样品溶液吸取与进样速度为50 µL/min. 此外,吐温-80在C18色谱柱上为强吸附,即使99%乙腈也难以洗脱完全,为保证色谱柱的快速清洗完全与分析重现性,本实验在色谱柱清洗阶段,在流动相中加入一定比例的异丙醇以提高流动相对色谱柱清洗能力,即25% B相(色谱纯乙腈),5% C相(超纯水)和70% D相(色谱纯异丙醇),清洗时间为20 min. 色谱柱清洗之后,色谱柱以色谱分离流动相平衡15 min. 样品溶液进样100 µL的色谱分离中,降解杂质峰出峰时间为25.0 min. 为避免因色谱柱超载使用导致的多次分离过程中杂质峰出峰时间漂移而影响降解杂质的萃取,本实验设定选择性模式(Select model)萃取21.0~25.0 min之间的洗脱液. 固相萃取柱为Spark公司的HySphereTM Resin GP(2*10 mm,23.6 mg)聚二乙烯苯树脂萃取柱. 萃取降解杂质之前,稀释泵(K-20 pump,Knaur)在液相色谱洗脱20.0 min时自动开启,水相以3 mL/min的流速泵入到色谱柱后的洗脱液中,稀释洗脱液中有机相比例以提高固相萃取柱对降解杂质的吸附能力. 实验设定50次自动在线固相萃取程序,实际运行33次萃取之后,软件意外终止实验. 萃取实验结束之后,以氮气吹干萃取降解杂质的固相萃取柱. NMR测试之前以氘代乙腈 (420 µL)洗脱萃取柱,并通过毛细管将萃取物的氘代乙腈溶液转移到安装于NMR磁体中的CryoFit插件流通池(60 µL),最后进行NMR检测. LC-DAD-SPE自动在线富集实验控制软件为Hystar 3.2.
1.3.3 NMR数据采集
本实验NMR数据采集流程与常规NMR实验完全相同. 一维1H NMR谱采用Topspin 3.5.7中脉冲序列库中标准单脉冲序列zg,采样点32 k,谱宽15 ppm,采样延迟2 s,谱中心设定为5.0 ppm,采样时间1.8 s,空扫次数为4,采样次数为32,实验时间约3 min. 1H-1H COSY谱采用标准序列cosygpmfqf,采样点2 k * 160,谱宽8.0 ppm,谱中心4.5 ppm,空扫次数为32,采样次数为2,采样延迟2 s,实验时间约 9 min. 1H-1H NOESY谱采用标准序列noesygpph,采样点为2 k * 144,谱宽8.0 ppm,谱中心4.5 ppm,空扫次数为16,采样次数为8,混合时间1 s,采样延迟2 s,实验时间约63 min. 1H-13C HSQC谱采用标准序列hsqcetgpsisp 2.2,采样点为2 k * 256,谱宽8.0 * 130 ppm,氢维中心4.7 ppm,碳维中心70.0 ppm,空扫次数为8,采样次数为2,采样延迟1.2 s,实验时间约13 min. 1H-13C HMBC谱采用标准序列库hmbcgpndqf,采样点为2 k * 360,谱宽10 * 205 ppm,氢维中心5.0 ppm,碳维中心107.5 ppm,空扫次数为8,采样次数为48,采样延迟1.0 s,本实验采用非均匀采样模式(NUS)采样,采样率为50%,实验时间约3 h. 以上NMR中,氢谱化学位移定标以氘代乙腈溶剂残余峰1.94 ppm定标,碳维以氘代乙腈中季碳117.8 ppm定标. 本研究中采用与上述相同的脉冲序列采集卡巴他赛的一维与二维NMR谱,其中1H-13C HMBC谱采样次数为8,以传统采样模式采集,实验时间为1 h.
2 结果与讨论
本研究采用LC-DAD-SPE-NMR/MS联用技术对卡巴他赛注射液样品中一个微量杂质进行了结构解析. 实验首先以LC-DAD-ESI-QTOF-MS模式对注射液样品进行分析,根据高分辨质谱数据确定未知杂质为卡巴他赛同分异构体,然后以LC-DAD-SPE-NMR模式萃取降解杂质后进行NMR实验,采集萃取物一维与二维NMR数据进行结构解析. 萃取物溶液1H NMR谱中谱峰复杂且重叠,最强峰来自吐温-80,且其强度高于降解杂质,掩盖了降解杂质部分谱峰,影响其直接识别. 本研究按混合物分析思路对采集的1H-1H COSY、1H-1H NOESY、1H-13C HSQC、1H-13C HMBC等二维谱进行细致而充分分析,在混合物谱峰中提取出降解杂质的氢-氢、氢-碳相关信息及氢、碳化学位移等信息,最终解析出降解杂质的化学结构.
采用LC-DAD-ESI-QTOF-MS对卡巴他赛注射液进行分析,得到卡巴他赛及未知降解杂质的色谱和质谱特征,判断降解杂质为卡巴他赛的同分异构体. 在230 nm液相色谱图中,卡巴他赛保留时间为13.0 min,而25.2 min色谱峰为降解杂质峰. 在质谱正离子模式下总离子流图谱中,在25.2 min色谱峰对应的一级质谱图中,出现一系列质量差为44 Da的离子峰,为典型含聚氧乙烯基化合物的质谱信号,结合注射液辅料成分种类,判断为吐温-80. 降解杂质与吐温-80同时从色谱柱上洗脱出,表明降解杂质与吐温-80疏水性相似,提示通过制备色谱方法制备高纯度降解杂质难度较大. 吐温-80离子化效率高,强烈抑制降解杂质的离子化,在正离子模式一级质谱中仅检测到低强度的降解杂质加钠离子峰m/z 858.3675(C45H57NO14Na,m/z理论值 858.3671)(图2).
图2
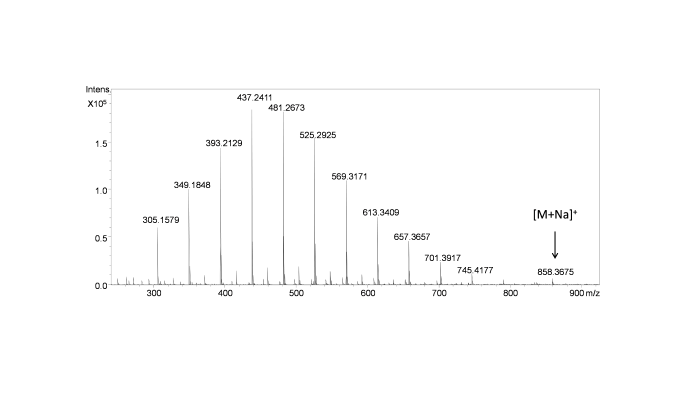
图2
卡巴他赛降解杂质一级质谱图
Fig. 2
MS spectrum of the degradation impurity of cabazitaxel
本研究进一步采用LC-DAD-SPE-NMR实验(图3)采集降解杂质NMR数据,提供更充分数据解析其结构. 自动萃取实验实际完成33次,仍成功萃取到足够量的降解杂质,采集出高质量的一维1H NMR与1H-1H COSY、1H-1H NOESY、1H-13C HSQC、1H-13C HMBC等二维NMR谱.
图3
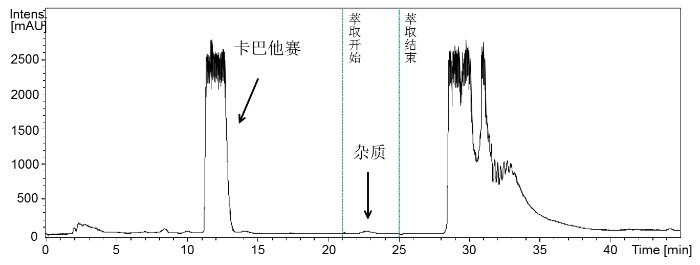
图3
卡巴他赛注射液降解杂质LC-DAD-SPE萃取色谱图
Fig. 3
LC-DAD-SPE trapping chromatogram of degradation impurity in cabazitaxel injection
对比卡巴他赛(图4(a))与萃取物(图4(b))的1H NMR谱,判断萃取物中含降解杂质与吐温-80,且吐温-80谱峰明显强度高于降解杂质,干扰降解杂质中高场区谱峰的直接识别. 萃取物1H NMR谱(图4(b))低场δH 7.30~8.20之间看到两组单取代苯环信号,与卡巴他赛化学信号结构中(图4(a))两个单取代苯环相近的化学位移和相似的峰形,判断为卡巴他赛降解杂质的特征信号. 萃取物1H NMR谱(图4(b))中δH 3.55峰强度异常高,高场δH 1.29处宽峰强度也较高,在1H-13C HSQC谱(图6)中与这两峰直接相连的碳化学位移为δC 70.1与29.0,分别为聚氧乙烯基团与脂肪链的特征信号,完全不同于卡巴他赛的谱峰特征(图4(a)),结合注射液辅料成分判断为吐温-80的谱峰. 评估1H NMR谱(图4(b))中降解杂质结构中H-2质子信噪比,以δH 5.59为信号,以δH 10.00 ~ 12.00区域为噪音计算信噪比(增宽因子为1 Hz)为1 127∶1,表明萃取物溶液中降解杂质浓度较为理想,并根据此信噪比设置了二维谱采样次数.
图4
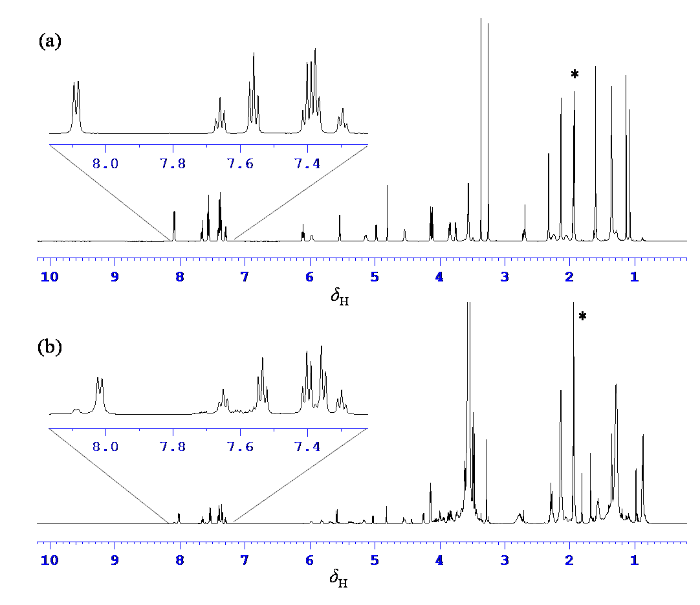
图4
(a)卡巴他赛与(b)降解杂质萃取物1H NMR谱(600 MHz,氘代乙腈,298 K). *:氘代乙腈溶剂残余峰
Fig. 4
1H NMR spectra of (a) cabazitaxel and (b) trapped analyte (600 MHz, in CD3CN, 298 K). * : residual peak of CD3CN
对萃取物的1H-1H COSY谱(图5)进行分析,可提取降解杂质中7个耦合系统中共25个氢原子的化学位移及连接关系.第一个耦合系统由δH 8.02(1H, d, 7.9 Hz)、7.65(2H, t, 7.4 Hz)和7.53(2H, t, 7.8 Hz)等5个质子组成,根据其化学位移和耦合特征判断这5个质子来自于一个单取代苯环. 第二个耦合系统同样由δH 7.43(2H, dd, 8.1, 7.4 Hz)、7.34(2H, t, 7.4 Hz)和7.30(1H, t, 7.4 Hz)等5个质子组成,根据其化学位移和耦合特征判断这些质子来自另一个单取代苯环. 第三个耦合系统由δH 5.98(1H, brd, 7.0 Hz)、5.17(1H, brd, 7.0 Hz)、4.53(1H, brs)和重叠的羟基δH 3.87等4个质子组成,与卡巴他赛化学结构中支链上4个质子化学位移和耦合特征相近. 第四个耦合系统由δH 5.82(1H, t, 7.4 Hz)和重叠的δH 2.00(1H)、2.17(1H)等3个质子组成,与卡巴他赛结构紫杉烷母核中环己烯环上三个质子化学位移和耦合特征相似. 第五个耦合系统由δH 5.59(1H, d, 7.7 Hz)与重叠的δH 3.58(1H)等2个质子组成,与卡巴他赛紫杉烷母核中八元环上相邻两个质子化学位移和耦合特征相似. 第六个耦合系统由δH 5.03(1H, d, 8.8 Hz)、3.86(1H, dd, 9.4, 7.4 Hz)、2.75(1H, ddd, 14.7, 8.8, 7.4 Hz)和重叠的δH 1.66(1H)等4个质子组成,与卡巴他赛紫杉烷母核中饱和六元环上4个质子化学位移和耦合特征相似. 第七个耦合系统由δH 4.26(1H, d, 8.1 Hz)与4.00(1H, d, 8.1 Hz)等2个质子组成,与卡巴他赛紫杉烷母核中含氧四元环连氧亚甲基质子化学位移和耦合特征相似. 根据以上25个质子组成的7个耦合系统分析,判断降解杂质与卡巴他赛化学结构高度相似.
图5
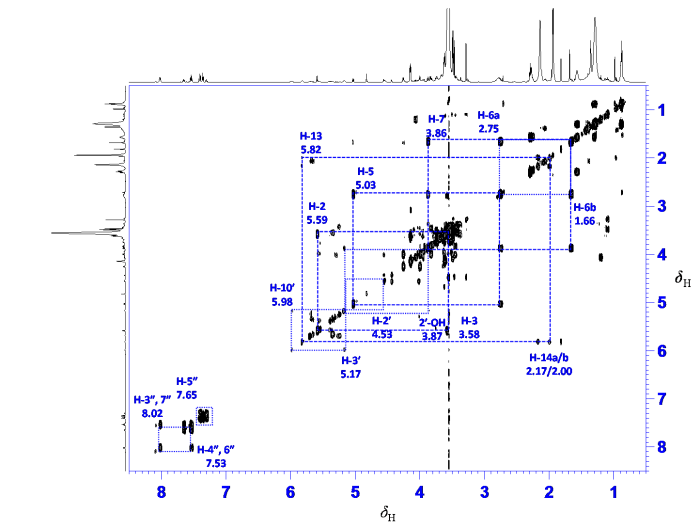
图5
萃取物1H-1H COSY谱及降解杂质氢-氢连接关系
Fig. 5
1H-1H COSY spectrum of the analyte and 1H-1H correlations in the degradation impurity
进一步分析萃取物1H-13C HSQC谱(图6),可指认降解杂质结构中7个耦合系统中25个质子直接相连的21个碳的化学位移. 与两个单取代苯环上10个质子直接相连的10个碳,在图6中可指认6个不同化学位移的碳(表1). 第三个耦合系统中2个质子δH 5.17(H-3′)、4.54(H-2′)分别与碳δC 56.6、74.0直接相连(表1),而两个质子δH 5.98(H-10′)、3.87(2′-OH)没有与之相连的碳,结合卡巴他赛谱峰分析(表1),指认δH 5.98为亚胺质子,δH 3.87为羟基质子. 第四个耦合系统中3个质子仅与两个碳直接相连,其中质子δH 2.17(H-14a)、2.00 (H-14b)与碳δC 35.6直接相连,质子δH 5.82(H-13)与碳δC 79.4直接相连. 第五个耦合系统中2个质子δH 5.59(H-2)、3.58(H-3)分别与碳δC 69.2、45.0直接相连. 第六个耦合系统中两个质子δH 5.03(H-5)、3.87(H-7)分别与碳δC 84.2、80.5直接相连,同时碳的化学位移表明这两个碳原子均连接氧原子;其中两个质子δH 2.75(H-6a)、1.66(H-6b)与同一个碳δC 33.6直接相连,表明此耦合系统中有一个亚甲基. 与第七个耦合系统中2个质子δH 4.26(H-20a)与4.00(H-20b)与一个碳δC 73.9直接相连. 通过1H-13C HSQC谱对降解杂质结构中21个碳化学位移指认,从碳化学位移的角度进一步证实降解杂质与卡巴他赛化学结构相似.
图6
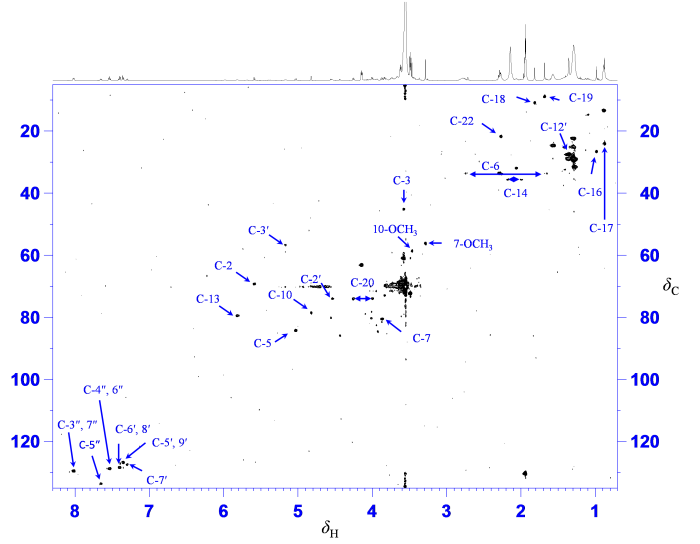
图6
萃取物1H-13C HSQC谱及降解杂质碳原子指认
Fig. 6
1H-13C HSQC spectrum of the analyte and the 13C assignment of the degradation impurity
表1 卡巴他赛与降解杂质的1H与13C NMR谱峰指认(600 MHz,氘代乙腈,298 K)
Table 1
| 卡巴他赛 | 降解杂质 | |||
|---|---|---|---|---|
| No. | δH | δC | δH | δC |
| 1 | 77.8 | 67.8 | ||
| 2 | 5.54 (d, 7.1 Hz) | 74.5 | 5.59 (d, 7.7 Hz) | 69.2 |
| 3 | 3.76 (d, 7.1 Hz) | 47.2 | 3.58 (overlaid) | 45.0 |
| 4 | 80.9 | 79.0 | ||
| 5 | 4.98 (d, 9.8 Hz) | 83.7 | 5.03 (d, 8.8 Hz) | 84.2 |
| 6 | 2.70 (ddd, 14.3, 9.8, 6.6 Hz) 1.61 (m, 14.3, 10.7, 2.2 Hz) | 31.9 | 2.75 (ddd, 14.7, 8.8, 7.4 Hz) 1.66 (overlaid) | 33.6 |
| 7 | 3.85 (dd, 10.7, 6.6 Hz) | 80.7 | 3.87 (dd, 9.4, 7.4 Hz) | 80.5 |
| 8 | 56.5 | 55.0 | ||
| 9 | 205.1 | 204.8 | ||
| 10 | 4.81 (s) | 82.5 | 4.82 (s) | 78.4 |
| 11 | 135.1 | 137.6 | ||
| 12 | 139.1 | 146.1 | ||
| 13 | 6.11 (td, 9.1, 1.1 Hz) | 71.5 | 5.82 (t, 7.4 Hz) | 79.4 |
| 14 | 2.25 (overlaid) 2.06 (overlaid) | 35.6 | 2.17 (overlaid) 2.00 (overlaid) | 35.6 |
| 15 | 43.1 | 74.4 | ||
| 16 | 1.13 (s) | 26.3 | 0.98 (s) | 26.6 |
| 17 | 1.08 (s) | 20.7 | 0.87 (s) | 24.0 |
| 18 | 1.61 (s) | 9.9 | 1.67 (s) | 8.9 |
| 19 | 1.93 (s) | 13.9 | 1.81 (s) | 11.0 |
| 20 | 4.15 (d, 8.3 Hz) 4.11 (d, 8.3 Hz) | 75.9 | 4.26 (d, 8.1 Hz) 4.00 (d, 8.1 Hz) | 73.9 |
| 21 | 170.3 | 169.9 | ||
| 22 | 2.33 (s) | 22.1 | 2.27 (s) | 21.8 |
| 1′ | 172.7 | * | ||
| 2′ | 4.53 (brs) | 73.8 | 4.54 (brs) | 74.0 |
| 3′ | 5.14 (brd, 7.8 Hz) | 56.8 | 5.17 (brd, 7.0 Hz) | 56.6 |
| 4′ | 139.4 | 139.4 | ||
| 5′, 9′ | 7.38 (dd, 8.0, 7.0 Hz) | 127.0 | 7.35 (d, 7.4 Hz) | 126.8 |
| 6′, 8′ | 7.40 (dd, 8.0, 7.1 Hz) | 128.4 | 7.40 (t, 7.4 Hz) | 128.5 |
| 7′ | 7.30 (dd, 7.1, 7.0 Hz) | 127.5 | 7.30 (t, 7.4 Hz) | 127.5 |
| 10′ | 5.97 (brd, 7.7 Hz) | 5.98 (brd, 7.0 Hz) | ||
| 11′ | * | * | ||
| 12′ | 78.9 | 78.9 | ||
| 13′ | 1.36 (s) | 27.5 | 1.35 (s) | 27.5 |
| 1″ | 165.7 | 165.8 | ||
| 2″ | 130.6 | 129.7 | ||
| 3″,7″ | 8.09 (d, 7.8 Hz) | 130.0 | 8.02 (d, 7.6 Hz) | 129.6 |
| 4″,6″ | 7.56 (t, 7.7 Hz) | 128. 6 | 7.53 (t, 7.6 Hz) | 128.8 |
| 5″ | 7.66 (t, 7.4 Hz) | 133.6 | 7.65 (t, 7.4 Hz) | 133.8 |
| 7-OCH3 | 3.26 (s) | 56.4 | 3.28 (s) | 56.1 |
| 10-OCH3 | 3.37 (s) | 56.2 | 3.46 (s) | 58.6 |
*本实验中未观测到. overlaid为与吐温-80谱峰重叠.
通过降解杂质1H-13C HMBC谱提供的氢-碳多键相关信息,可确定耦合系统中质子,以及甲基与甲氧基等孤立质子与临近多个碳的连接关系,可进一步解析降解杂质化学平面结构. 第一个耦合系统中单取代苯环上质子δH 8.02(H-3″,7″)与苯环上碳δC 129.6(C-3″,7″)、133.8(C-5″)及环外季碳δC 165.8(C-1″)有多键相关,季碳δC 165.8化学位移可能为酯键碳,因此推测结构中苯甲酸酯基团. 第二个耦合系统中单取代苯环上质子δH 7.35(H-5′,9′)与苯环外碳δC 56.6(C-3′)有多键相关,而碳δC 56.6(C-3′)来自于第三个耦合系统,因此推测苯环第二个单取代苯环连接在第三个耦合系统中C-3′位. 孤立甲基δH 1.81(H-19)与碳原子δC 79.4(C-13)、137.6(C-11)、146.1(C-12),其中季碳δC 137.6(C-11)与146.1(C-12)化学位移提示为典型双键碳的特征,表明甲基(H-19)连接在双键上. 同时此甲基与碳原子δC 79.4(C-13)的远程连接,可确定其连接在C-12位. 单峰δH 4.82(H-10)与碳原子δC 58.5(10-OMe)、67.8(C-1)、137.6(C-11)、146.1(C-12)及羰基碳δC 204.8(C-9)有远程连接,而甲氧基质子δH 3.46(10-OMe)与叔碳δC 78.4(C-10)有远程连接,可确定此甲基δH 4.82连接在C-10位,且有甲氧基δH 3.46连接在与其直接相连的叔碳(C-10)上. 甲基单峰δH 1.67(H-18)与δC 45.0(C-3)、55.0(C-8)、80.5(C-7)及羰基碳δC 204.8有远程相关,可确定此甲基连接在C-8位. 叔碳δC 80.5(C-7)还与甲氧基单峰δH 3.28有远程连接,提示此甲氧基连接在C-7位,而叔碳质子δH 3.87(H-7)与甲氧基碳δC 56.1的远程连接可进一步证实甲氧基的连接位置. 与叔碳质子δH 3.87(H-7)相邻的亚甲基质子δH 2.75(H-6a)、1.66(H-6b)与δC 55.0(C-8)、79.0(C-4)、80.5(C-7)、84.2(C-6)等碳有远程连接,叔碳质子δH 5.03(H-5)与邻近碳δC 79.0(C-4)与80.5(C-7)有远程连接,叔碳质子δH 3.58(H-4)与邻近四个碳δC 55.0(C-8)、67.8(C-1)、79.0(C-4)与80.5(C-7)有远程连接,这些氢-碳远程连接符合紫杉烷母核中六元环的结构特征. 亚甲氧基质子δH 4.26(H-20a)、4.00(H-20b)与邻近碳δC 45.0(C-3)、79.0(C-4)与84.2(C-5)有远程连接,与卡巴他赛紫杉烷母核中含氧四元环相似的特征. 叔碳质子δH 5.59(H-4)与邻近四个碳δC 35.6(C-14)、45.0(C-3)、55.0(C-8)、67.8(C-1)、74.4(C-15)有远程连接,可确定此质子的位置.
分析降解杂质中两个季碳δC 67.8(C-1)、74.4(C-15)的远程相关时,发现与卡巴他赛结构中对应的两个季碳δC 77.8(C-1)和43.1(C-15)化学位移有显著差别,因此推测这两个碳的解析是降解杂质结构解析的关键. 在降解杂质1H-13C HMBC谱局部(图7(a)),可观察到其中甲基质子δH 0.98(H-16)与碳δC 24.0(C-17)、67.8(C-1)和74.4(C-15)的远程相关,和另一个甲基质子δH 0.87(H-17)和碳δC 26.6(C-16)、67.8(C-1)和74.4(C-15)的远程相关. 而卡巴他赛1H-13C HMBC谱中(图7(d)),可观察到甲基质子δH 1.13(H-16)与碳δC 21.3(C-17)、77.8(C-1)、43.1(C-15)和135.6(C-11)的远程连接,和另一个甲基质子δH 1.08(H-17)和碳δC 27.0(C-16)、77.8(C-1)、43.1(C-15)和135.6(C-11)的远程连接. 杂质中与卡巴他赛中季碳δC 135.6(C-11)对应的季碳δC 137.6(C-11),并未观察到其与两个甲基δH 0.98(H-16)、0.87(H-17)的远程连接关系,提示这两甲基与环上双键距离较远,与目前已知的卡巴他赛相关杂质有显著差别,判断为卡巴他赛相关杂质研究中未见文献报道的新杂质. 进一步查找文献,日本和中国学者曾从红豆杉属植物中分离出多个重排的紫杉烷类化合物[25⇓-27],重排后的产物中C-15化学位移向低场移动到75 ppm附近[25,27],而C-1化学位移向高场有一定移动,其中重排产物taxchinin A的结构还被X-射线单晶衍射的确认[25]. 文献[27]中chinentaxunine中两个重排的甲基质子与碳的远程连接关系,与本文中杂质的重排基团上两个甲基质子δH 0.98(H-16)和0.87(H-17)的远程连接关系一致. 结合降解杂质的NMR特征及文献[25⇓-27],确认降解杂质结构中紫杉烷母核发生重排,解析出降解杂质的结构(图1). 文献[28]报道紫杉醇在酸性条件下会发生重排,紫杉烷母核中不饱和六元环发生重排变成不饱和五元环,环上偕二甲基碳重排,以异丙醇基连接在C-1位,因此推测此降解杂质产生机制可能为酸性条件下发生碳正离子重排.
图7
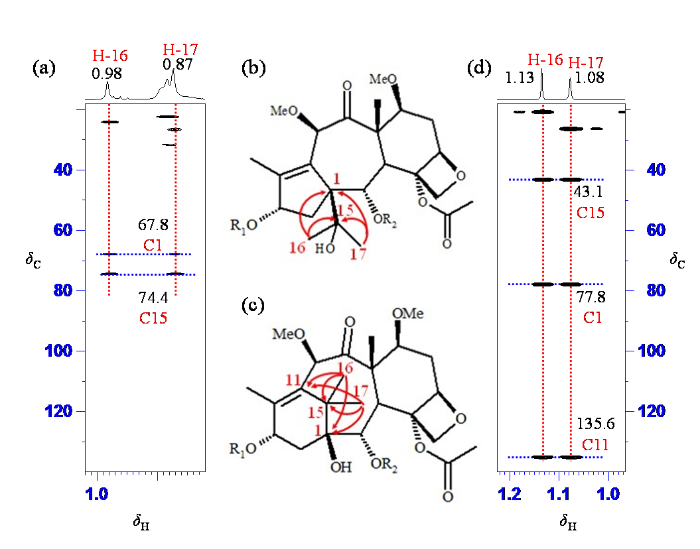
图7
降解杂质(a) 1H-13C HMBC谱局部及(b)关键氢-碳多键相关;卡巴他赛(d) 1H-13C HMBC谱局部及(c)关键氢-碳多键相关
Fig. 7
Part of 1H-13C HMBC spectra and key 1H-13C multiple-bond correlations of degradation impurity [(a), (b)] and cabazitaxel [(d), (c)]
本研究进一步通过对比降解杂质与卡巴他赛1H-13C HMBC谱中紫杉烷母核与其上支链及取代基的远程相关特征,确定了降解杂质支链及取代基没有发生变化. 降解杂质1H NMR谱中支链上三个质子δH 5.98(H-10′)、5.17(H-3′)、4.54(H-2′)峰形较宽,在1H-13C HMBC谱中没有观察到与这三质子远程连接的碳. 卡巴他赛结构中对应的质子δH 5.97(H-10′)、5.15(H-3′)、4.53(H-2′)峰形同样较宽,1H-13C HMBC谱中未观察到与这三质子远程连接.结合卡巴他赛NMR谱学研究文献[29],及对比卡巴他赛与降解杂质化学结构中支链上氢-碳相关信号及化学位移特征(表1),判断支链结构没有发生变化. 卡巴他赛1H NMR谱中紫杉烷母核质子δH 6.11(H-13)峰形较好,在1H-13C HMBC谱中可观察到其与支链上酯键碳δC 172.7(C-1′)的远程相关. 而降解杂质与之对应的质子δH 5.82(H-13)在1H NMR谱中为宽的(伪)三重峰,在1H-13C HMBC谱中未找到与任何碳的远程连接关系,同时在NOE谱中也未观察到该质子与支链质子的NOE相关信号,通过化学位移比对确定支链与结构母核的连接位置. 降解杂质中叔丁氧基中甲基质子δH 1.36(H-13′),在1H-13C HMBC谱中仅观察到其与叔丁氧基内的两种δC 27.5(C-13′)、78.9(C-12′)有远程相关,未观察到与酰胺碳(C-11′)的远程相关. 卡巴他赛中对应的叔丁氧基中质子δH 1.36(H-13′),在1H-13C HMBC谱中同样只观察到其与δC 27.5(C-13′)、78.9(C-12′)的远程连接,未见与酰胺碳(C-11′)的远程相关. 降解杂质中乙酰基质子δH 2.27(H-22)在1H-13C HMBC谱中与酰基碳δC 169.9存在远程相关. 卡巴他赛中对应的酰基质子δH 2.33(H-22)在1H-13C HMBC谱中同样只与酰基碳δC 170.2存在远程相关,两者中均未观察到酰基质子与紫杉烷母核中碳(C-4)的远程连接. 根据以上分析,指认降解杂质中1H、13C NMR谱峰见表1.
对杂质的1H-1H NOESY谱进行分析,推测杂质X中重排基团与chinentaxunine中重排基团相同的构型[27]. 降解杂质的构型主要通过重排后的异丙醇基中两个甲基(H-16与H-17)与五元环上确定构型的H-13的NOE关系来判断. 在NOE谱(图8)中甲基δH 0.98(H-16)与δH 2.17(H-14a)、5.59(H-2)和5.82(H-13)等质子存在NOE相关,甲基δH 0.87(H-17)与δH 2.17(H-14a)、和5.59(H-2)等质子存在NOE相关. 其中甲基质子δH 0.98(H-16)与5.82(H-13)存在NOE相关,因此判断重排后的异丙醇基为R构型. 此外,在NOE谱中可观察到第二个苯环上质子δH 7.35(H-5′, 9′)与支链上质子δH 5.98(H-10′)、5.17(H-3′)、4.54(H-2′)的相关,进一步证实第二个耦合系统和第三个耦合系统连接在一起.
图8
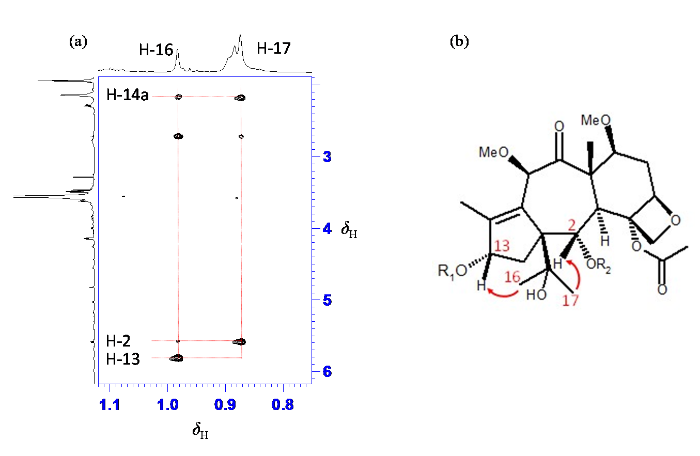
图8
降解杂质(a) 1H-1H NOESY谱局部及(b)关键氢-氢NOE相关
Fig. 8
(a) Part of 1H-1H NOESY spectra and (b) key NOE correlations of degradation impurity
3 结论
本研究中卡巴他赛注射液中未知杂质含量约为0.3% ~ 0.4%,其疏水性与辅料吐温-80相近,采用液相色谱分离制备方法难以分离得到高纯度降解杂质. 本研究采用LC-DAD-SPE-NMR/MS联用技术对该杂质进行了分析,成功解析出降解杂质结构. 实验过程通过两种运行模式采集MS和NMR数据,以LC-DAD-ESI-QTOF-MS工作模式通过高分辨质谱数据确定降解杂质高分辨MS数据,以LC-DAD-SPE-NMR工作模式对降解杂质色谱峰进行多次固相萃取后采集NMR数据. 萃取物1H NMR谱中含高比例的辅料吐温-80的信号,严重干扰降解杂质NMR谱峰直接识别,本研究以混合物分析模式,借助1H-1H COSY、1H-1H NOESY、1H-13C HSQC、1H-13C HMBC等二维NMR谱提供的氢-氢、氢-碳相关信息,得到降解杂质氢、碳化学位移,成功排除吐温-80导致谱峰的干扰,最终鉴定出化学结构.
与传统方法相比,采用LC-DAD-SPE-NMR/MS 联用技术表现出诸多优势:大大减少样品与试剂消耗,更为环保;自动化程度高,大大减少制备杂质所需要的劳动量;效率高,在较短时间内完成复杂的实验.
本项目采用LC-DAD-SPE-NMR/MS联用技术对卡巴他赛注射液中微量杂质进行了分析,尽管萃取出的降解杂质中混杂高比例的吐温-80,本研究仍能鉴定出该降解杂质的化学结构,表明联用技术对复杂样品体系的微量成分强大分析能力,且在药物杂质研究领域具有广阔的应用前景.
利益冲突
无
致谢
降解杂质结构解析得到云南大学化学科学与工程学院卢崇道教授指点和帮助.
参考文献
Impurities in New Drug Substances Q3A(R2)
[R],
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Impurities in New Drug Products Q3B(R2)
[R],
Application of a trap-free two-dimensional liquid chromatography combined with ion trap/time-of-flight mass spectrometry for separation and characterization of impurities and isomers in cefpiramide
[J].
Mass spectrometry for small molecule pharmaceutical product development: A review
[J].
DOI:10.1002/mas.20289
PMID:21500245
[本文引用: 1]

Developing a pharmaceutical product has become increasingly difficult and expensive. With an emphasis on developing project knowledge at an earlier stage in development, the use of information-rich technologies (particularly MS) has continued to expand throughout product development. Continued improvements in LC/MS technology have widened the scope of utilizing MS methods for performing both qualitative and quantitative applications within product development. This review describes a multi-tiered MS strategy designed to enhance and accelerate the identification and profiling of both process- and degradation-related impurities in either the active pharmaceutical ingredient (API) or formulated product. Such impurities can be formed either during chemical synthesis, formulation, or during storage. This review provides an overview of a variety of orthogonal-mass spectrometric methodologies, namely GC/MS, LC/MS, and ICP-MS, in support of product development. This review is not meant to be all inclusive; however, it has been written to highlight the increasing use of hyphenated MS techniques within the pharmaceutical development area.Copyright © 2010 Wiley Periodicals, Inc.
Mass spectrometry-based structure elucidation of small molecule impurities and degradation products in pharmaceutical development
[J].
Identification of pharmaceutical impurities
[J].
LC-NMR-MS in drug discovery
[J].Nuclear magnetic resonance spectroscopy (NMR) is arguably the most versatile analytical platform for complex mixture analysis. Specifically, interfacing liquid chromatography with parallel NMR and mass spectrometry (LC-NMR-MS) gives comprehensive structural data on metabolites of novel drugs in development. Applications in natural product, combinatorial chemistry and drug metabolism studies are reviewed. Microcoil probes and capillary separation methods have enormous potential. Recent innovations to improve NMR detection limits include CryoFlowProbes and on-line solid-phase extraction (LC-SPE-NMR). These state-of-the-art analytical platforms are widely applicable to identifying novel candidate drugs from diverse complex mixtures within a drug discovery strategy.
Critical investigation of coupled liquid chromatography-NMR spectroscopy in pharmaceutical impurity identification
[J].
LC-UV-Solid-Phase Extraction-NMR-MS combined with a cryogenic flow probe and its application to the identification of com-pounds present in greek oregano
[J].
Hyphenation of solid-phase extraction with liquid chro-matography and nuclear magnetic resonance: Application of HPLC-DAD-SPE-NMR to identification of constituents of Kanahia laniflora
[J].
Identification of three novel polyphenolic compounds, origanine A-C, with unique skeleton from Origanum vulgare L. using the hyphenated LC-DAD-SPE-NMR/MS methods
[J].
HPLC-SPE-NMR in pharmaceutical development: capabilities and applications
[J].
Hyphenated NMR methods in natural products research, part 2: HPLC-SPE-NMR and other new trends in NMR hyphenation
[J].This review describes the principles and performance of a novel and highly promising hyphenated technique, HPLC-SPE-NMR, which is based on post-column analyte trapping by solid-phase extraction. The analytes are subsequently eluted from the SPE cartridges using deuterated solvents. This indirect HPLC-NMR hyphenation offers numerous advantages compared to direct HPLC-NMR methods. Multiple trapping leads to a dramatic increase of analyte amounts available for NMR, enabling acquisition of high-quality 2D NMR data within a short time. Other new developments, including combination of solenoidal coil capillary flow-probes with microflow HPLC, are also discussed. Fast extract dereplication using these techniques enables focusing of isolation efforts on truly novel and promising natural products, based on precise structural data obtained with crude extracts or fractions.
Important roles of the hyphenated HPLC-DAD-MS-SPE-NMR technique in metabonomics
[J].
LC-DAD-MS/SPE-NMR hyphenation. A tool for the analysis of pharmaceutically used plant extracts: Identification of iso-baric iridoid glycoside regioisomers from Harpagophytum procumbens
[J]
Minor nortriterpenoids from the twigs and leaves of Rhododendron latoucheae
[J].
The use of LC/MS, GC/MS, and LC/NMR hyphenated techniques to identify a drug degradation product in pharmaceutical development
[J].Understanding drug degradation in the formulated product is critical in pharmaceutical development as it has significant impacts on drug efficacy, safety profile and storage conditions. As a result, identification of degradation compounds has taken an important role in the drug development process. In this study, various hyphenated analytical techniques, such as liquid chromatography mass spectrometry (LC/MS), gas chromatography mass spectrometry (GC/MS), and liquid chromatography nuclear magnetic resonance with a solid phase extraction interface (LC/SPE/NMR), have been applied to the identification of a drug degradation product which grew over time in the stability study of the drug product. The target unknown is less polar and more unsaturated than the drug substance based upon reverse phase HPLC relative retention time and UV spectra. It is not ionizable by electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) in either a positive or a negative mode. The unknown was isolated by an HPLC fraction collector and enriched by solid phase extraction. GC/MS with chemical ionization (CI) was employed to determine the molecular weight of this compound. Its fragmentation pattern was determined by CI-MS/MS using an ion trap mass spectrometer. The isolated material was also analyzed by LC/SPE/NMR, from which the structure of this compound was further characterized. The study utilizes a combination of various hyphenated analytical techniques to obtain complimentary information for structure elucidation of the unknown. The combination approach is critical for unambiguous impurity structure elucidation in drug degradation studies of pharmaceutical drug products.
Semi-preparative LC-SPE-cryoflow NMR for impurity identifications: use of mother liquor as a better source of impurities
[J].
Rapid identification of unknown impurities in 3-Bromo-5-(trifluoromethyl) aniline by LC-SPE/NMR
[J].
Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer
[J].
DOI:10.2147/DDDT.S13029
PMID:21448449
[本文引用: 1]
Until recently, patients with castration-resistant prostate cancer (CRPC) had limited therapeutic options once they became refractory to docetaxel chemotherapy, and no treatments improved survival. This changed in June 2010 when the Food and Drug Administration (FDA) approved cabazitaxel as a new option for patients with CRPC whose disease progresses during or after docetaxel treatment. For most of these patients, cabazitaxel will now replace mitoxantrone (a drug that was FDA-approved because of its palliative effects) as the treatment of choice for docetaxel-refractory disease. The approval of cabazitaxel was based primarily on the TROPIC trial, a large (n = 755) randomized Phase III study showing an overall median survival benefit of 2.4 months for men with docetaxel-pretreated metastatic CRPC receiving cabazitaxel (with prednisone) compared to mitoxantrone (with prednisone). Cabazitaxel is a novel tubulin-binding taxane that differs from docetaxel because of its poor affinity for P-glycoprotein (P-gp), an ATP-dependent drug efflux pump. Cancer cells that express P-gp become resistant to taxanes, and the effectiveness of docetaxel can be limited by its high substrate affinity for P-gp. Preclinical and early clinical studies show that cabazitaxel retains activity in docetaxel-resistant tumors, and this was confirmed by the TROPIC study. Common adverse events with cabazitaxel include neutropenia (including febrile neutropenia) and diarrhea, while neuropathy was rarely observed. Thus, the combination of cabazitaxel and prednisone is an important new treatment option for men with docetaxel-refractory metastatic CRPC, but this agent should be administered cautiously and with appropriate monitoring (especially in men at high risk of neutropenic complications).
Isolation, identification and characterization of potential impurities in cabazitaxel and their formation
[J].
Development and validation of a stability-indicating HPLC Method for the determination of the impurities in cabazitaxel
[J].
A validated stability indicating UPLC method for simultaneous determination of related substances, and degradation products of cabazitaxel drug substance and its pharmaceutical injection forms
[J].
Taxchinin a: A diterpenoid from Taxus chinensis
[J].
A rearranged taxane from the himalayan yew Taxus wallichiana
[J].
Taxane diterpenoids from the seeds of Chinese yew Taxus chinensis
[J].
Acid catalyzed conversions of toxoids
[J].
Spectroscopic studies and structural elucidation of cabazitaxel
[J].
卡巴他赛结构确证的波谱学研究
[J].
DOI:10.11938/cjmr20170208
[本文引用: 1]
用红外吸收光谱、紫外吸收光谱、核磁共振(NMR)波谱(包含<sup>1</sup>H NMR、<sup>13</sup>C NMR、DEPT-135、COSY、HSQC和HMBC)、质谱、差示扫描量热分析、X-射线粉末衍射和元素分析等方法对卡巴他赛原料药精制品进行了结构确证,对其所有的<sup>1</sup>H和<sup>13</sup>C NMR信号进行了归属,同时讨论了质谱主要碎片离子可能的裂解方式和红外特征吸收峰所对应的官能团的振动形式,通过多种波谱技术结合确证了卡巴他赛的结构.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}