


引言
细胞色素c(cytochrome c,cyt c)是一个分子量为12.5 kDa的水溶性蛋白质,通常定位于真核生物的线粒体. 在线粒体中,cyt c是电子传递链(electron transport chain,ETC)的重要组成部分,它通过分子中血红素辅基(heme)中心的铁原子在二价(Fe2+)和三价(Fe3+)之间的转换实现电子传递[1],这就导致cyt c可产生两种不同的状态,即还原态构象和氧化态构象[2⇓-4]. 如图1所示,cyt c还原态和氧化态的两种构象结构相似,都呈球状结构,包含由Ω-loop连接的5个α-螺旋,以及两个很短的β-折叠,heme共价结合在多肽链上,两种构象之间可以发生可逆的转换[5].
图1

图1
氧化态(青色)和还原态(紫色)cyt c的晶体结构(PDB ID分别为2YCC和1YCC)
Fig. 1
The crystal structure of Fe3+ cyt c (cyan) and Fe2+ cyt c (purple) (PDB ID: 2YCC and 1YCC)
但是当细胞受到外界刺激,比如受高渗透压、高盐等刺激时,cyt c会被释放到胞质中,进而启动细胞凋亡程序[6]. 虽然cyt c从线粒体释放是不可逆的,但有研究[7]表明,在cyt c释放到胞质后,细胞凋亡的执行是受其高度调控的,在脱氧腺苷三磷酸(deoxyadenosine triphosphate,dATP)存在时,cyt c会与胞质中的凋亡蛋白酶活化因子1(apoptosis peptide enzyme activation factor 1,Apaf-1)结合形成凋亡小体复合物,并激活含半胱氨酸的天冬氨酸蛋白水解酶(cysteinyl aspartate specific proteinase, caspase),进而启动细胞凋亡. 然而,cyt c的氧化态和还原态具有不同的理化性质,虽然关于它们是否都能够激活凋亡小体进而启动细胞凋亡的研究备受关注,但目前结果仍然存在争议[5]. 有研究分别使用健康和凋亡的细胞匀浆进行实验,结果发现在健康的人急性T淋巴细胞白血病细胞(Jurkat cells)的匀浆中,cyt c被细胞质中的还原型谷胱甘肽、过氧化氢酶、硫氧还蛋白等还原性物质迅速还原,并可能起到抑制细胞凋亡的作用[8]. 而在凋亡的人宫颈癌细胞(HeLa cells)中,由于线粒体外膜的通透性变大,cyt c会被同时释放到胞质中的细胞色素氧化酶迅速氧化,而且只有氧化态的cyt c才能激活凋亡小体[9]. 这表明释放到胞质中的cyt c的氧化还原状态可能会发生改变,只有氧化态的cyt c才能启动细胞凋亡. 但是另一些研究[10]发现无论是氧化态还是还原态的cyt c都可以与Apaf-1结合形成凋亡小体,即cyt c可能不需要改变氧化还原状态就可以激活细胞凋亡途径.
在凋亡时cyt c会获得很高的过氧化物酶活性[11]. 在其他的过氧化物酶中,heme通常以五配位的状态存在[12];而正常情况下在cyt c中处于六配位,His18是heme的轴向配体,Met80是其远端配体,从而形成一个Fe-Met80六配位的结构,heme被深埋在蛋白中,无法与溶剂接触,使其处于过氧化物酶活性很低的状态[13]. 因此,cyt c释放到胞质后,可能同时也涉及到heme的配位改变. cyt c在释放到胞质中的氧化还原状态和heme配位状态产生争议的可能原因是蛋白构象识别的研究中常用的紫外-可见吸收光谱(ultraviolet-visible absorption spectrum,UV-Vis)易受背景影响[8]. 尽管由于铁原子三价和二价特征不同,通过观测UV-Vis光谱中不同波段的特征吸收峰,很容易区分氧化态和还原态的cyt c[14]. 但是细胞内复杂环境导致的cyt c可能产生的构象变化[13]在UV-Vis光谱中可能并不能呈现出很明显的变化.
核磁共振(nuclear magnetic resonance,NMR)是研究生物大分子(蛋白质、DNA等)结构及动力学的理想工具,并且可在原子分辨率水平表征蛋白质的构象变化[15⇓-17]. 但有研究[18]将酿酒酵母iso-1 cyt c突变体进行15N标记,尝试获取它在大肠杆菌细胞匀浆中的构象变化,结果却发现样品的1H-15N HSQC信号严重衰减甚至消失,这可能是由于cyt c与匀浆中某些成分之间存在一定的弱相互作用. 而基于甲基的NMR检测可用于超大分子量蛋白、蛋白复合体或者复杂环境中蛋白质结构的研究. 这是因为相比于主链上的残基,侧链上的甲基更加灵活,具有更长的横向弛豫时间,有利于NMR信号的获取[19,20].
1 实验部分
1.1 实验材料
野生型iso-1 cyt c(纯度≥85%)、重水(D2O)均购自Sigma-Aldrich公司;葡萄糖、无水磷酸氢二钠(Na2HPO4)、无水磷酸二氢钠(NaH2PO4)、氯化钠(NaCl)、铁氰化钾(K3[Fe(CN)6])、过氧化氢(H2O2)、抗坏血酸钠(L-ascorbic acid sodium salt,SLA)购于国药集团化学试剂有限公司,均为分析纯;酵母粉(Yeast extract)、胰蛋白胨(Tryptone)购自Oxoid公司;酿酒酵母BY4741菌株购自ThermoFisher公司.
1.2 野生型iso-1 cyt c的纯化及不同构象的cyt c稀溶液样品制备
野生型iso-1 cyt c在使用前按照文献[23]报道的两步法,先后采用HiTrap SP HP阳离子交换色谱柱(GE Healthcare)和HiLoad Superdex 75 pg制备尺寸排阻色谱柱(GE Healthcare)进行了纯化,随后液氮速冻保存于-80 ℃,使用时将其溶解在20 mmol/L的Na2HPO4 / NaH2PO4缓冲液(PB,pH 7.0)中. 纯化后的野生型iso-1 cyt c通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE)和质谱鉴定纯度为95%以上,可用于后续实验.
稀溶液样品制备:在野生型iso-1 cyt c溶液中分别加入0.5 mmol/L K3[Fe(CN)6]、SLA和H2O2得到天然氧化态、天然还原态以及五配位构象的cyt c,加入相应试剂后先在1.5 mL离心管内上下翻转10 min,再静置15 min,使其充分接触和反应,由于K3[Fe(CN)6]和SLA有一定的紫外吸收,需要用3 kDa浓缩管将样品中残留的K3[Fe(CN)6]和SLA除去,H2O2不需除去. 再将cyt c稀释到合适的浓度,UV-Vis实验所用的浓度为0.02 mmol/L,NMR实验所用的浓度为0.1 mmol/L(含10% D2O用于锁场).
1.3 酵母细胞的培养和cyt c-细胞匀浆混合样品的制备
酿酒酵母细胞在YEPD培养基(含1%的酵母粉、2%的胰蛋白胨、2%的葡萄糖)中培养48 h至OD600值达到2左右. 3 000 g离心5 min收集细胞沉淀,并用20 mmol/L PB(pH 7.0)重悬,使用高压裂菌仪进行破碎并收集细胞匀浆液,并通过BCA蛋白定量试剂盒(碧云天生物)测定其总蛋白浓度.
将冻干的cyt c样品溶于总蛋白浓度为20 mg/mL的酵母细胞匀浆中,使cyt c终浓度为1 mmol/L. 样品中需要加入10% D2O用于锁场.
1.4 UV-Vis光谱实验
UV-Vis光谱实验在SpectraMax i3x 酶标仪(Molecular Devices)上进行,在96孔板上加入约100 μL浓度为0.02 mmol/L的cyt c溶液,检测波长范围为300~600 nm.
1.5 NMR实验和数据处理
NMR实验在配备TXI低温探头的Bruker Avance 700 MHz NMR谱仪上进行的,实验温度为25 ℃. 2D 1H-13C HSQC使用hsqcetgp脉冲序列,F2(1H)和F1(13C)维的谱宽分别为11 363.64 Hz和7 043.37 Hz,谱中心分别为3 291.18 Hz和9 332.13 Hz. 1H维和13C维的采样数据点阵为t2×t1=2 048×64,累加次数64. 实验数据采用Bruker Topspin 4.0.1软件进行处理.
2 结果与讨论
2.1 不同构象cyt c稀溶液样品的UV-Vis光谱和NMR谱图
本文首先对不同构象cyt c蛋白稀溶液样品的UV-Vis光谱和NMR谱图特征进行了比较. 我们根据文献[14,22],分别将野生型iso-1 cyt c处理成还原态、氧化态、以及五配位构象,进而观测其谱学特征,结果如图2所示. 在过量弱还原剂SLA存在时,cyt c的中心铁原子为Fe2+,Fe-Met80配位键未断裂(标记为“Fe2+-Met80”构象),此时在UV-Vis光谱的522 nm和550 nm处出现了两个特征吸收峰[图2(a)][24];而在NMR谱中出现位于δH 2.85的Lys72me3信号[图2(e)]. 在过量弱氧化剂K3[Fe(CN)6]存在时,cyt c的中心铁原子为Fe3+,Fe-Met80配位键未断裂(标记为“Fe3+-Met80”构象),此时在UV-Vis光谱529 nm处出现了特征吸收峰[图2(b)][24],但是相比于“Fe2+-Met80”构象的吸收峰,该吸收峰明显变宽而且变弱,不易辨认;而在NMR谱图δH 3.31处出现Lys72me3的信号[图2(f)],该信号远离“Fe2+-Met80”构象相应基团的信号. 由此可见,在区分cyt c在稀溶液中的两种天然构象时,虽然灵敏度低,但由于两种状态下Lys72me3的化学位移差别明显,因此相比UV-Vis光谱,NMR更易用于区分该蛋白的构象,及观测其变化.
图2
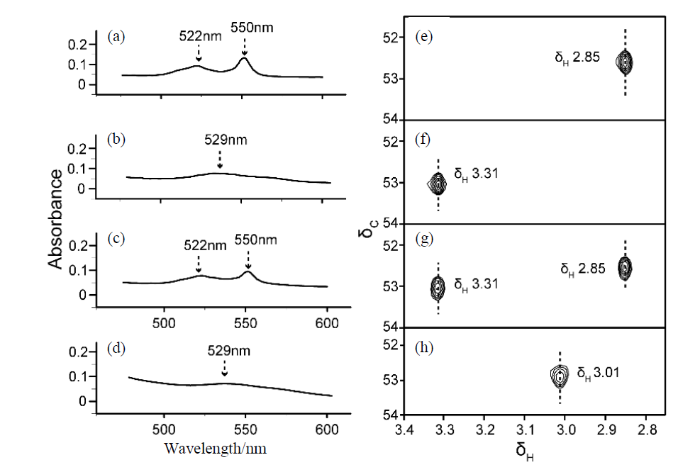
图2
不同构象cyt c的UV-Vis光谱和NMR谱. (a)经SLA处理过的cyt c、(b)经K3[Fe(CN)6]处理过的cyt c、(c)分别经SLA和K3[Fe(CN)6]处理过的cyt c等比例混合、(d)经H2O2处理过的cyt c的UV-Vis光谱;(e)经SLA处理过的cyt c、(f)经K3[Fe(CN)6]处理过的cyt c、(g)分别经SLA和K3[Fe(CN)6]处理过的cyt c等比例混合、(h)经H2O2处理过的cyt c的2D 1H-13C HSQC谱图
Fig. 2
UV-Vis absorption spectra and NMR spectra of cyt c in different conformations. UV-Vis spectra of (a) cyt c treated with SLA, (b) cyt c treated with K3[Fe(CN)6], (c) cyt c mixed in equal proportions treated with SLA and K3[Fe(CN)6], respectively, (d) cyt c treated with H2O2; 2D 1H-13C HSQC spectra of (e) cyt c treated with SLA, (f) cyt c treated with K3[Fe(CN)6], (g) cyt c mixed in equal proportions treated with SLA and K3[Fe(CN)6], respectively, (h) cyt c treated with H2O2
为了进一步分析不同构象的谱学特征,我们将“Fe2+-Met80”构象和“Fe3+-Met80”构象的cyt c以摩尔比1:1混合,然后分别采集它的UV-Vis光谱与NMR谱图[图2(c)和2(g)]. 可以看出,在UV-Vis光谱上,522 nm和550 nm的特征吸收峰信号较为明显,但529 nm处的信号几乎无法辨认,难以确定该处是否有吸收峰存在[图2(c)]. 根据文献[25],此时只能通过监测550 nm处特征吸收峰的强度变化来反映其还原态构象的相对含量变化. 而在NMR谱中,δH 2.85和3.31处同时出现两个明显的信号[图2(g)],分别与还原构象和氧化构象对应,且两者的相对信号强度基本相同. 这一结果也表明NMR相比UV-Vis光谱更适用于多种混合状态下cyt c构象的检测,可简单迅捷的获得蛋白构象及其相对含量的变化信息.
cyt c在H2O2存在的情况下,原本的Fe-Met80配位键将会断裂,进而形成五配位的heme,此外,由于H2O2的氧化性,cyt c中心的铁原子为Fe3+,因此,将其标记为“Fe3+-”构象[14]. 在UV-Vis光谱中,该构象仅可观测到529 nm处的吸收峰[图2(d)],表明其铁原子为Fe3+,但无法看出heme的配位是否发生了变化. 而在NMR谱中,δH 2.85和3.31处的两个信号都消失,取而代之的是在δH 3.01处出现了一个新的信号[图2(h)]. 则这个信号代表的正是“Fe3+-”构象[14]. 这表明NMR除了可以表征cyt c的氧化态和还原态,还可以检测其与heme配位变化有关的其他构象.
综上所述,UV-Vis光谱和NMR技术均可用于观测cyt c的氧化还原构象. UV-Vis光谱虽然操作简便、灵敏度高、样品用量少,但难以区分拥有五配位heme的cyt c其他构象. 而由于不同状态cyt c蛋白修饰甲基的化学位移差别大,NMR技术更易区分氧化态、还原态乃至不同heme配位的cyt c构象,可用于复杂的细胞匀浆环境中cyt c构象变化的检测.
2.2 cyt c在细胞匀浆中的构象变化追踪
细胞内成分复杂,而且氧化还原性质与稀溶液不同,这些因素可能导致cyt c在细胞匀浆中的构象与其在稀溶液中有所差异. 因此,我们通过将纯化的iso-1 cyt c与细胞匀浆均匀混合,以它的Lys72me3为天然探针,采用2D 1H-13C HSQC技术,追踪了cyt c在酵母细胞匀浆中的构象变化,结果如图3所示.
图3

图3
cyt c在酵母细胞匀浆液中不同时间点的2D 1H-13C HSQC谱图
Fig. 3
2D 1H-13C HSQC spectra of cyt c in yeast cell homogenate at different time points
cyt c通常是氧化态和还原态共存的,在加入到酵母细胞匀浆液中后,也可明显看到这两种状态的存在. 在加入到酵母细胞匀浆液1 h时,很容易从谱上观察到有三个信号:位于δH 3.31和2.85处的信号分别为cyt c原始的“Fe3+-Met80”构象和“Fe2+-Met80”构象,而位于δH 3.11处的信号经空白对照实验证明是来自于酵母细胞匀浆中的背景信号,根据化学位移将其归属为胆碱(Choline)的特征甲基. 随着时间的推移,这些信号的相对强度开始发生变化. 在第2 h时,δH 3.38和2.98处分别出现了新的信号,这两个信号均是由于铁原子的配位发生变化而产生的,铁原子与Met80的配位键断裂,又与其他残基重新配位.根据配位的残基差异以及文献[22]报道,δH 3.38的信号为取代了Fe3+-Met80配位而形成Fe3+-Lys73配位的cyt c构象(标记为“Fe3+-Lys73”构象).相关研究[26]表明当用盐酸胍处理cyt c时,它会变成双His配位的构象,并且只有当His33缺失时,His26才参与heme配位. 而在过量盐酸胍存在时,Lys72me3在NMR谱上化学位移正好为δH 2.98,因此,我们观察到δH 2.98的信号可能主要是Fe-His33配位的构象,并且铁原子为Fe3+,将其标记为“Fe3+-His33”构象. 据报道相较“Fe3+-Met80”构象,这两种配位的构象结构更松散,因而具有较高过氧化酶活性[27],会在细胞经历氧化应激时存在[28]. 在酵母细胞匀浆环境中,这些信号随着时间的推移出现,可能是由于匀浆的长时间放置,里面的活性氧(reactive oxygen species,ROS)会逐渐增加,导致氧化应激的发生[29]. 由于“Fe3+-Lys73”构象和“Fe3+-His33”构象的cyt c具有较高的过氧化物酶活性,因此这些构象的出现表明cyt c可能参与了ROS的清除,以高过氧化酶活性的状态抵抗氧化应激. 在第3h时,可以看到在δH 3.01处又出现了一个新的信号,该信号是Fe-Met80的配位键断裂,以五配位heme形式存在的构象,为“Fe3+-”构象[14]. 这意味着随着时间的增长,ROS的积累增加,导致“Fe3+-Lys73”和“Fe3+-His33”构象的cyt c配位键进一步断裂,cyt c的构象向“Fe3+-”构象转变,它的过氧化物酶活性达到最大. 此外,可以看到,随着时间的进一步增加,虽然没有新的信号出现,但出现的五个来自不同cyt c构象信号的相对强度发生了改变.
图4为所观测信号相对强度变化曲线. 从图中很容易看出,随着cyt c在细胞匀浆中的时间增长,原始氧化态的蛋白,即“Fe3+-Met80”构象的cyt c相对含量在逐渐减少,而“Fe2+-Met80”构象、“Fe3+-Lys73”构象、“Fe3+-His33”构象和“Fe3+-”构象的cyt c的含量在逐渐增加. 同时,观测到的来自匀浆的Choline信号强基本保持不变,表明增强的信号均来自cyt c本身的构象变化.
图4

图4
cyt c的不同构象在酵母细胞裂解液中的含量随时间变化曲线
Fig. 4
Content changes of cyt c with different conformations in yeast cell homogenate over time
根据文献报道,细胞质内是一个还原环境,胞质内的cyt c可以被还原型谷胱甘肽、过氧化氢酶、硫氧还蛋白等还原[8],同时也可以被H2O2等ROS物质和氧化酶氧化[9]. 但目前还不清楚这些酶的作用在细胞匀浆中的强弱. 实验结果表明:加入到细胞匀浆后,cyt c的“Fe3+-Met80”构象减少,而“Fe2+-Met80”构象增多的原因可能是一部分cyt c受匀浆内各种还原物质的作用,发生了还原. 但是“Fe3+-Lys73”、“Fe3+-His33”构象和“Fe3+-”构象的出现可能是一部分cyt c在其他ROS物质的作用下,无法被还原,且致使Fe-Met80键断开,cyt c发生构象转换. “Fe3+-Lys73”和“Fe3+-His33”构象使cyt c结构变的较为松散,因此可进一步从六配位heme构象展开为五配位heme构象,使得heme暴露程度不断变大,获得最大的过氧化物酶活性,更好的清除ROS,保护细胞免受氧化应激的伤害. 前期的研究中我们在稀溶液中做过H2O2的滴定实验模拟ROS增加,随着H2O2浓度的增加,cyt c构象的变化趋势为紧凑的六配位状态(Fe3+/2+-Met80)→较松散的六配位状态(Fe3+-His33)→展开的五配位状态(Fe3+-),对应的过氧化物酶活性逐渐变强. 与本研究中观测到的现象是相吻合的,表明细胞匀浆中增加的ROS物质诱导cyt c的构象朝着具有较高过氧化物酶活性的状态转变.
3 结论
NMR技术可在原子分辨率水平探测蛋白构象,本文通过NMR实验追踪野生型iso-1 cyt c侧链修饰甲基的化学位移变化,检测了野生型iso-1 cyt c在酵母细胞匀浆液中的构象变化,发现细胞匀浆对cyt c的构象存在很大影响,cyt c在细胞匀浆中至少存在4种不同的氧化态构象和1种还原态构象,而且随着时间的推移,匀浆中的环境变化可导致cyt c不同构象之间发生相互转换. 这一发现表明cyt c的构象会随环境改变,构象的改变可能与细胞抵抗ROS刺激密切相关. 甲基可以作为一个有效的探针用于探测蛋白质在复杂细胞环境中的结构变化.
致谢
感谢国家重点基础研究发展计划(2017YFA0505400,2018YFE0202300,2018YFA0704002)的支持.
利益冲突
无
参考文献
Electronic connection between the quinone and cytochrome c redox pools and its role in regulation of mitochondrial electron transport and redox signaling
[J].
DOI:10.1152/physrev.00006.2014
PMID:25540143
[本文引用: 1]

Mitochondrial respiration, an important bioenergetic process, relies on operation of four membranous enzymatic complexes linked functionally by mobile, freely diffusible elements: quinone molecules in the membrane and water-soluble cytochromes c in the intermembrane space. One of the mitochondrial complexes, complex III (cytochrome bc1 or ubiquinol:cytochrome c oxidoreductase), provides an electronic connection between these two diffusible redox pools linking in a fully reversible manner two-electron quinone oxidation/reduction with one-electron cytochrome c reduction/oxidation. Several features of this homodimeric enzyme implicate that in addition to its well-defined function of contributing to generation of proton-motive force, cytochrome bc1 may be a physiologically important point of regulation of electron flow acting as a sensor of the redox state of mitochondria that actively responds to changes in bioenergetic conditions. These features include the following: the opposing redox reactions at quinone catalytic sites located on the opposite sides of the membrane, the inter-monomer electronic connection that functionally links four quinone binding sites of a dimer into an H-shaped electron transfer system, as well as the potential to generate superoxide and release it to the intermembrane space where it can be engaged in redox signaling pathways. Here we highlight recent advances in understanding how cytochrome bc1 may accomplish this regulatory physiological function, what is known and remains unknown about catalytic and side reactions within the quinone binding sites and electron transfers through the cofactor chains connecting those sites with the substrate redox pools. We also discuss the developed molecular mechanisms in the context of physiology of mitochondria. Copyright © 2015 the American Physiological Society.
Three-dimensional solution structure of Saccharomyces cerevisiae reduced iso-1-cytochrome c
[J].Two-dimensional 1H NMR spectra of Saccharomyces cerevisiae reduced iso-1-cytochrome c have been used to confirm and slightly extend the assignment available in the literature. 1702 NOESY cross-peaks have been assigned, and their intensities have been measured. Through the program DIANA and related protocols (Güntert, 1992), a solution structure has been obtained by using 1442 meaningful NOEs and 13 hydrogen-bond constraints. The RMSD values with respect to the mean structure for the backbone and all heavy atoms for a family of 20 structures are 0.61 +/- 0.09 and 0.98 +/- 0.09 A, the average target function value being as small as 0.57 A2. The larger number of slowly exchanging amide NHs observed in this system compared to that observed in the cyanide derivative of oxidized Ala 80 cytochrome c suggests that the oxidized form is much more flexible and that the backbone protons are more solvent accessible. Comparison of the present structure with the crystal structures of reduced yeast cytochrome c and of the complex between cytochrome c peroxidase and oxidized yeast cytochrome c reveals substantial similarity among the backbone conformations but differences in the residues located in the region of protein-protein interaction. Interestingly, in solution the peripheral residues involved in the interaction with cytochrome c peroxidase are on average closer to the position found in the crystal structure of the complex than to the solid state structure of the isolated reduced from.
Solution structure of oxidized saccharomyces cerevisiae iso-1-cytochrome c
[J].The solution structure of oxidized Saccharomycescerevisiae Cys102Ser iso-1-cytochromechas been determined using 1361 meaningful NOEs (of 1676 total) after extending the published proton assignment [Gao, Y., et al. (1990) Biochemistry 29, 6994-7003] to 77% of all proton resonances. The NOE patterns indicate that secondary structure elements are maintained upon oxidation in solution with respect to the solid state and solution structures of the reduced species. Constraints derived from the pseudocontact shifts [diamagnetic reference shift values are those of the reduced protein [Baistrocchi, P., et al. (1996) Biochemistry 35, 13788-13796]] were used in the final stages of structure calculations. After restrained energy minimization with constraints from NOEs and pseudocontact shifts, a family of 20 structures with rmsd values of 0.58 +/- 0.08 and 1.05 +/- 0.10 A (relative to the average structure) for the backbone and all heavy atoms, respectively, was obtained. The solution structure is compared with the crystal structure and the structures of related systems. Twenty-six amide protons were detected in the NMR spectrum 6 days after the oxidized lyophilized protein was dissolved in D2O (pH 7.0 and 303 K); in an analogous experiment, 47 protons were observed in the spectrum of the reduced protein. The decrease in the number of nonexchanging amide protons, which mainly are found in the loop regions 14-26 and 75-82, confirms the greater flexibility of the structure of oxidized cytochrome c in solution. Our finding of increased solvent accessibility in these loop regions is consistent with proposals that an early step in unfolding the oxidized protein is the opening of the 70-85 loop coupled with dissociation of the Met80-iron bond.
Conformational change of wild type cytochrome c characterized by NMR spectroscopy at natural isotropic abundance
[J].
天然同位素丰度野生型酵母细胞色素c构象变化的核磁共振检测
[J].
Regulation of apoptosis by the redox state of cytochrome c
[J].DOI:10.1016/j.bbabio.2008.03.024 URL [本文引用: 2]
Apoptosis in yeast
[J].
DOI:10.1016/j.mib.2004.10.012
PMID:15556039
[本文引用: 1]
Apoptosis is a highly regulated cellular suicide program crucial for metazoan development. However, dysfunction of apoptosis also leads to several diseases. Yeast undergoes apoptosis after application of acetic acid, sugar- or salt-stress, plant antifungal peptides, or hydrogen peroxide. Oxygen radicals seem to be key elements of apoptotic execution, conserved during evolution. Furthermore, several yeast orthologues of central metazoan apoptotic regulators have been identified, such as a caspase and a caspase-regulating serine protease. In addition, physiological occurrence of cell death has been detected during aging and mating in yeast. The finding of apoptosis in yeast, other fungi and parasites is not only of great medical relevance but will also help to understand some of the still unknown molecular mechanisms at the core of apoptotic execution.
Cytochrome c: functions beyond respiration
[J].
Cytochrome c is rapidly reduced in the cytosol after mitochondrial outer membrane permeabilization
[J].
DOI:10.1007/s10495-010-0455-2
PMID:20094799
[本文引用: 3]
Visible spectroscopy was used to measure real-time changes in the oxidation state of cytochrome c (cyt c) and the a-cytochromes (cyt aa(3)) of cytochrome oxidase during mitochondrial outer membrane permeabilization (MOMP) initiated by anisomycin in HL-60 cells. The oxidation state of mitochondrial cyt c was found to be approximately 62% oxidized before MOMP and became approximately 70% oxidized after MOMP. In contrast, the cytosolic pool of cyt c was found to be almost fully reduced. This oxidation change allows cyt c release to be continuously and quantitatively monitored in real time. Anoxia and antimycin were used to fully reduce and fully oxidize, respectively, the mitochondrial pool of cyt c and it was found that the release of cyt c was independent of it oxidation state consistent with a simple model of cyt c passively diffusing down a concentration gradient through a pore or tear in the outer membrane. After MOMP was complete, the flux of cyt c diffusing back into the mitochondria was measured from the residual mitochondrial oxygen consumption after complete inhibition of the bc(1) with antimycin and myxothiazol. The outer membrane was found to be highly permeable after MOMP implying that the reduction of cyt c in the cytosol must be very rapid. The permeability of the outer membrane measured in this study would result in the release of cyt c with a time constant of less than 1 s.
Mitochondrial regulation of caspase activation by cytochrome oxidase and tetramethylphenylenediamine via cytosolic cytochrome c redox state
[J].
DOI:10.1074/jbc.M700322200
PMID:17690099
[本文引用: 2]
Cytochrome c release from mitochondria induces caspase activation in cytosols; however, it is unclear whether the redox state of cytosolic cytochrome c can regulate caspase activation. By using cytosol isolated from mammalian cells, we find that oxidation of cytochrome c by added cytochrome oxidase stimulates caspase activation, whereas reduction of cytochrome c by added tetramethylphenylenediamine (TMPD) or yeast lactate dehydrogenase/cytochrome c reductase blocks caspase activation. Scrape-loading of cells with this reductase inhibited caspase activation induced by staurosporine. Similarly, incubating intact cells with ascorbate plus TMPD to reduce intracellular cytochrome c strongly inhibited staurosporine-induced cell death, apoptosis, and caspase activation but not cytochrome c release, indicating that cytochrome c redox state can regulate caspase activation. In homogenates from healthy cells cytochrome c was rapidly reduced, whereas in homogenates from apoptotic cells added cytochrome c was rapidly oxidized by some endogenous process. This oxidation was prevented if mitochondria were removed from the homogenate or if cytochrome oxidase was inhibited by azide. This suggests that permeabilization of the outer mitochondrial membrane during apoptosis functions not just to release cytochrome c but also to maintain it oxidized via cytochrome oxidase, thus maximizing caspase activation. However, this activation can be blocked by adding TMPD, which may have some therapeutic potential.
Oxidized or reduced cytochrome c and axial ligand variants all form the apoptosome in vitro
[J].DOI:10.1021/acs.biochem.7b00309 URL [本文引用: 1]
Becoming a peroxidase: Cardiolipin-induced unfolding of cytochrome c
[J].DOI:10.1021/jp402104r URL [本文引用: 1]
Horseradish peroxidase: a modern view of a classic enzyme
[J].Horseradish peroxidase is an important heme-containing enzyme that has been studied for more than a century. In recent years new information has become available on the three-dimensional structure of the enzyme and its catalytic intermediates, mechanisms of catalysis and the function of specific amino acid residues. Site-directed mutagenesis and directed evolution techniques are now used routinely to investigate the structure and function of horseradish peroxidase and offer the opportunity to develop engineered enzymes for practical applications in natural product and fine chemicals synthesis, medical diagnostics and bioremediation. A combination of horseradish peroxidase and indole-3-acetic acid or its derivatives is currently being evaluated as an agent for use in targeted cancer therapies. Physiological roles traditionally associated with the enzyme that include indole-3-acetic acid metabolism, cross-linking of biological polymers and lignification are becoming better understood at the molecular level, but the involvement of specific horseradish peroxidase isoenzymes in these processes is not yet clearly defined. Progress in this area should result from the identification of the entire peroxidase gene family of Arabidopsis thaliana, which has now been completed.
Alternative conformations of cytochrome c: Structure, function, and detection
[J].
DOI:10.1021/acs.biochem.5b01385
PMID:26720007
[本文引用: 2]
Cytochrome c (cyt c) is a cationic hemoprotein of ∼100 amino acid residues that exhibits exceptional functional versatility. While its primary function is electron transfer in the respiratory chain, cyt c is also recognized as a key component of the intrinsic apoptotic pathway, the mitochondrial oxidative protein folding machinery, and presumably as a redox sensor in the cytosol, along with other reported functions. Transition to alternative conformations and gain-of-peroxidase activity are thought to further enable the multiple functions of cyt c and its translocation across cellular compartments. In vitro, direct interactions of cyt c with cardiolipin, post-translational modifications such as tyrosine nitration, phosphorylation, methionine sulfoxidation, mutations, and even fine changes in electrical fields lead to a variety of conformational states that may be of biological relevance. The identification of these alternative conformations and the elucidation of their functions in vivo continue to be a major challenge. Here, we unify the knowledge of the structural flexibility of cyt c that supports functional moonlighting and review biochemical and immunochemical evidence confirming that cyt c undergoes conformational changes during normal and altered cellular homeostasis.
Cytochrome c as a peroxidase: Activation of the precatalytic native state by H2O2-induced covalent modifications
[J].DOI:10.1021/jacs.7b07106 URL [本文引用: 5]
Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48
[J].
Monitoring the conformational flexibility of cytochrome c at low ionic strength by 1H-NMR spectroscopy
[J].DOI:10.1046/j.1432-1327.1998.2560271.x URL [本文引用: 1]
Visualizing the pre-active conformation of response regulator PhoBNF20D in its apo state
[J].
探测应答调控蛋白PhoBNF20D自由态中存在的Pre-Active构象
[J].
Protein interactions in the Escherichia coli cytosol: an impediment to in-cell NMR spectroscopy
[J].DOI:10.1002/cbic.201100063 URL [本文引用: 1]
The dynamic process of beta(2)-adrenergic receptor activation
[J].DOI:10.1016/j.cell.2013.01.008 URL [本文引用: 1]
Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins
[J].
DOI:10.1016/j.sbi.2015.03.009
PMID:25881211
[本文引用: 1]
Nuclear magnetic resonance (NMR) spectroscopy is a uniquely powerful tool for studying the structure, dynamics and interactions of biomolecules at atomic resolution. In the past 15 years, the development of new isotopic labeling strategies has opened the possibility of exploiting NMR spectroscopy in the study of supra-molecular complexes with molecular weights of up to 1MDa. At the core of these isotopic labeling developments is the specific introduction of [(1)H,(13)C]-labeled methyl probes into perdeuterated proteins. Here, we describe the evolution of these approaches and discuss their impact on structural and biological studies. The relevant protocols are succinctly reviewed for single and combinatorial isotopic-labeling of methyl-containing residues, and examples of applications on challenging biological systems, including high molecular weight and membrane proteins, are presented. Copyright © 2015 Elsevier Ltd. All rights reserved.
Determinants of cytochrome c pro-apoptotic activity - the role of lysine 72 trimethylation
[J].DOI:10.1074/jbc.275.21.16127 URL [本文引用: 1]
Monitoring alkaline transitions of yeast iso-1 cytochrome c at natural isotopic abundance using trimethyllysine as a native NMR probe
[J].DOI:10.1039/C8CC07605G URL [本文引用: 3]
Expression of doubly labeled Saccharomyces cerevisiae iso-1 ferricytochrome c and H-1,C-13 and N-15 chemical shift assignments by multidimensional NMR
[J].DOI:10.1016/S0014-5793(00)02032-9 URL [本文引用: 1]
Stability of uniformly labeled (13C and 15N) cytochrome c and its L94G mutant
[J].DOI:10.1038/s41598-021-86332-w URL [本文引用: 2]
Importance of the redox state of cytochrome c during caspase activation in cytosolic extracts
[J].DOI:10.1042/bj3290095 URL [本文引用: 1]
Identification of the predominant non-native histidine ligand in unfolded cytochrome c
[J].The heme and its two axial ligands, His18 and Met80, play a central role in the folding/unfolding mechanism of cytochrome c. Because of the covalent heme attachment, His18 remains bound under typical denaturing conditions, while the more labile Met80 ligand is replaced by an alternate histidine ligand. To distinguish between the two possible non-native histidine ligands in horse cytochrome c, variants with a His26 to Gln or His33 to Asn substitution were prepared using a yeast expression system. Protonation of the non-native histidine ligand in the GuHCl-denatured state results in a pronounced blue shift of the Soret heme absorbance band (low-spin to high-spin transition). While substitution of His26 has no effect on the apparent pKa of this transition (5.7 +/- 0.05), the H33N variant exhibits a substantially higher pKa (6.1 +/- 0.05), indicating that His33 is the dominant sixth heme ligand in denatured cytochrome c and that His26 (or another nitrogenous group) acts as a ligand in the absence of a histidine at position 33. The kinetics of the pH-induced ligand dissociation shows two phases which were assigned to each of the two histidine ligands on the basis of their distinct temperature dependence. Despite their nearly identical equilibrium unfolding transitions, the two histidine mutants show differences in their folding kinetics. While the kinetic behavior of H26Q cyt c is very similar to that of the wild-type, the H33N mutation leads to loss of a kinetic phase with a rate in the 2-10 s-1 range that has previously been attributed to the rate-limiting dissociation of a trapped non-native histidine, which is thus identified as His33.
pH dependence of formation of a partially unfolded state of a Lys 73→His variant of iso-1-cytochrome c: Implications for the alkaline conformational transition of cytochrome c
[J].DOI:10.1021/bi0017778 URL [本文引用: 1]
Lysine carbonylation is a previously unrecognized contributor to peroxidase activation of cytochrome c by chloramine-T
[J].DOI:10.1039/C8SC03624A URL [本文引用: 1]
Mitochondrial oxidative stress in aging and healthspan
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}