


引言
耦合常数(Coupling constant)和NOE(Nuclear Overhauser effect)效应是局部特性的NMR参数,能够推测大部分有机小分子和生物大分子的结构信息,是天然产物立体构型确定最常用的方法.近年来,NOE效应的定量使用在天然产物构型研究中也展现出独特的优势.但是它们通常提供的是分子中相距较近的片段信息,对于空间距离较远的手性片段或者多季碳手性片段,其相应构型的确定有时候也存在不确定性[9].
偶极耦合是两个磁性核(即自旋核)通过空间传递而产生的一种非常重要的相互作用[9,15].然而,在固体NMR谱图中,分子的偶极耦合作用太强而导致谱图分辨率差且谱峰过宽;在液体NMR谱图中,分子的偶极耦合作用反而会因为分子受到的布朗运动而被完全平均为零,使得偶极耦合无法被进一步的研究和探索[15].残留偶极耦合(Residual dipolar coupling,RDC)是在溶液中分子的运动被约束时,其偶极耦合可能没有被完全平均,从而保留的较小的偶极耦合残留值[16].RDC作为一种非局部特性NMR参数和NMR各向异性参数,能够提供长程或者全局性的独特空间信息,而不是局限于空间距离较近的短程结构信息,其大小与核间距及其键矢量与外加静磁场之间的相对取向相关,能够反映分子结构的整体构造、正确构型以及优势构象[14⇓⇓-17].RDC的数据计算如(1)式所示:
西松烷二萜类化合物是一类具有14元大环骨架的柔性大环分子,结构新颖独特,主要来源于短指软珊瑚(Sinularia)、肉芝软珊瑚(Sarcophyton)与豆荚软珊瑚(Lobophyton)三种软珊瑚属[14,25-26].西松烷二萜类化合物存在丰富的手性中心,其中C-C单键的自由旋转导致了构象的数目庞大和预测困难问题.利用NOE和量子化学计算等方法确定该类化合物的立体构型会存在错误的情况[27].因此,本文以从软珊瑚Sarcophyton elegans分离得到的西松烷二萜类化合物sinulariol Z[28]为代表,首次采用RDC确定其立体构型,并通过单晶X射线衍射法确证其立体构型,再次验证了RDC在西松烷二萜类化合物立体构型确定方面的可行性.
1 实验部分
1.1 仪器与试剂
实验所用的薄层色谱硅胶(GF254)购于青岛海洋化工有限公司;柱色谱硅胶(100~200目,200~300目)购于青岛海洋化工厂分厂;葡聚糖凝胶(SNphadex LH-20)购于GE healthcare公司;石油醚、甲醇等分析纯试剂购于天津博迪化工股份有限公司;定向介质AAKLVFF多肽(纯度超过98%)购于希施生物科技(上海)有限公司.
旋转蒸发仪型号为Heizbad HB digit(德国Heidolph公司);高效液相色谱仪型号为Waters 2695(美国Waters公司);分析型高效液相色谱柱型号为Agilent Extend-C18: 250 mm×6 mm,5 μm(美国Agilent公司);半制备型高效液相色谱柱型号为YMC-Pack Pro C18: 250 mm×10 mm,5 μm(日本YMC株式会社).
单晶数据采集于Bruker APEX-ⅡCCD衍射仪.
1.2 样品提取与分离
软珊瑚Sarcophyton elegans样品于2018年采自南海西沙岛鸭公岛海域,样品编号为XS-18-yg-114,存放于中国海洋大学医药学院海洋馆天然产物一室,由台湾国立海洋生物博物馆,海洋生物研究所宋秉钧教授鉴定为Sarcophyton elegans.将冷冻保存的Sarcophyton elegans样品(湿重为7.9 kg)剪碎,于室温条件下用甲醇提取6次(每次3天),经过滤得到滤液后在真空中浓缩蒸干,再经过无水甲醇溶解脱盐,得到粗浸膏178.0 g.粗浸膏经硅胶柱层析,洗脱剂为石油醚-丙酮(洗脱梯度为400:1~1:1)体系和二氯甲烷-甲醇(洗脱梯度为20:1~1:1)体系,得到14个一级部位(Fr 1~Fr 14),再采用薄层色谱法对各组分进行分析.其中Fr 7经硅胶柱层析,洗脱剂为石油醚-丙酮(洗脱梯度为50:1~1:1)体系,得到6个二级部位(Frs.7.1~Frs.7.6).通过高效液相色谱,以MeOH-H2O系统(75:35)洗脱,从Frs.7.6中分离得到化合物1(2.5 mg).
1.3 NMR实验参数
1H NMR在Bruker Avance 400 MHz NMR波谱仪,以CDCl3为溶剂,采用标准脉冲程序采集,观测频率为400.13 MHz,谱宽为8 196.7 Hz,累加次数32;13C NMR在JNM-ECZ600R/S1 600 MHz NMR波谱仪上完成,以CDCl3为溶剂,采用标准脉冲程序采集,观测频率为150.92 MHz,谱宽为37 878.8 Hz,累加次数478;各向同性(MeOD-d4)和各向异性(AAKLVFF多肽)条件下的perfect 1JCH -resolved HSQC在Bruker Avance 400 MHz NMR波谱仪上完成,使用脉冲序列Perfect-1JCH-HSQC[29]采集,F2(H)和F1(C)维谱宽分别为8 196.7 Hz和2 999.4 Hz,采集数据点阵t1*t2=2 048*256,累加次数26.NMR实验温度为298.2 K.
1.4 实验RDC值获取
为了获取实验RDC值,本文将待测化合物扩散于定向介质中,给溶质分子提供一个各向异性环境.该环境约束了分子运动,使分子在磁场中呈现一定的定向排列,从而保证偶极耦合作用没有被完全抵消.定向介质是提取化合物RDC数据的关键,本文选用的定向介质是AAKLVFF多肽[15,18].随后,进行了perfect 1JCH-resolved HSQC二维NMR实验,分别测量了待测化合物(2.0~5.0 mg)在各向同性(MeOD-d4)和各向异性(AAKLVFF多肽)条件下perfect 1JCH-resolved HSQC谱图,得到了待测化合物在各向同性环境中标量耦合常数(nJ)以及在各向异性环境中的复合耦合值(nT,即标量耦合和偶极耦合之和).实验RDC值(1DCH值)则是两者之间的差值[9].
1.5 理论RDC值获取
RDC计算数值是由Maestro V11.9(Schrödinger Inc.)软件[30]、高斯09程序包[31]以及MSpin软件[32]结合得到的.首先是化合物所有可能的相对构型的构象获取,通过Maestro V11.9(Schrödinger Inc.)软件中MacroModel的Conformational Search程序模块,对化合物所有可能的非对映异构体进行构象搜索.设置分子力场为OPLS3e,以GB/SA Octanol为溶剂模型,能量阈值为10 kJ mol-1,并采用均方根偏差(Root-mean-squared-distance,RMSD,截断值为0.5 Å)以及Mixed Torsional/Low-Mode sampling(MTLMOD,最大迭代次数为2 500次)以消除冗余构象.通过Maestro V11.9(Schrödinger Inc.)软件中MacroModel的Minimization程序模块,采用与构象搜索相同的RMSD、MTLMOD参数设置以及能量阈值,对所得构象进行初步优化并去除不稳定构象.随后,采用密度泛函理论(DFT),基于B3LYP/6-31G(d,p)理论水平,选取初步优化后获得的所有构象,在高斯09程序包中进行进一步优化和振动频率分析(附件图S1和表S1).各非对映异构体中构象的玻尔兹曼分布比例则是通过计算得到的吉布斯自由能结合玻尔兹曼分布率计算得到的(附件表S2).最终,经MacroModel/Maestro将得到的每个相对构型的构象(玻尔兹曼分布比例大于1%)进行叠加后,导入Mspin软件中采用单张近似法预测得到理论RDC值.
1.6 RDC数据分析
将实验RDC值导入Mspin软件中,采用奇异值分解(Singular value decomposition,SVD)拟合预测[9],使实验测得的RDC值与预测得到的理论RDC值进行匹配拟合,从而得到质量因子(Q因子).基于上述各非对映异构体中构象(玻尔兹曼分布比例大于1)的玻尔兹曼分布比例,采用fit population设定(温度为298.15 K),Mspin软件能够计算得到各非对映异构体的构象各自的Q因子,再根据构象占比得到各个相对构型最终Q因子.Q因子是实验RDC值和预测理论RDC值的均方根,用于评估分子的构型在二者之间表现出来的一致性,Q因子值越接近于零,表明通过计算模拟预测的构型与实际构型越一致[33,34].
2 结果与讨论
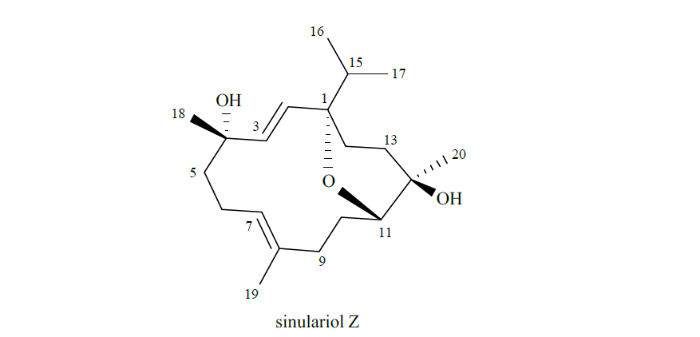
2.1 Sinulariol Z的结构解析
经过一维NMR实验,得到了化合物1的1H NMR和13C NMR数据(谱图见附件图S2和S3).将化合物1的NMR数据与文献中的数据对比,得出该化合物为sinulariol Z[28].化合物sinulariol Z最初是从我国南海软珊瑚Sinularia rigida中分离得到.通过分析其红外光谱、1H NMR谱图、13C NMR谱图以及HSQC谱图,推测其结构特征为一个西松烷二萜类化合物,再根据1H-1H COSY谱中连续质子相关信号构建出的结构片段,HMBC谱图相关信号,以及高分辨质谱和不饱和度,佐证了化合物sinulariol Z的平面结构(图1)[28]. 随后,通过与其结构类似的化合物sinulariol J和sinulariol X对比NMR谱图数据,确定该化合物的立体构型为1S、4S、11S、12R[28].然而,仅利用NMR数据确定化合物的立体构型存在风险,因此本文通过RDC参数和单晶X射线衍射法再次确定了该化合物的立体构型.
图1
2.2 Sinulariol Z的立体结构确定
将化合物sinulariol Z分别在各向同性和各向异性环境中测得的perfect 1JCH-resolved HSQC谱图进行覆盖叠加,得到图2.图2中,红色等高线为该化合物在各向同性中的perfect 1JCH-resolved HSQC信号,蓝色等高线为该化合物在各向异性环境中的perfect 1JCH-resolved HSQC信号,红色框区域为该化合物在两种条件下的关键信号的覆盖叠加图及其放大图.基于该化合物的perfect 1JCH-resolved HSQC覆盖叠加图(图2),分别测定得到该化合物在各向同性条件下的标量耦合常数(nJ)以及在各向异性条件下的复合耦合值(nT),通过计算两者的差值得到1DCH值(1DCH = nT -nJ),即实验RDC值.最终,共提取了化合物sinulariol Z的12组实验RDC值(表1).
图2
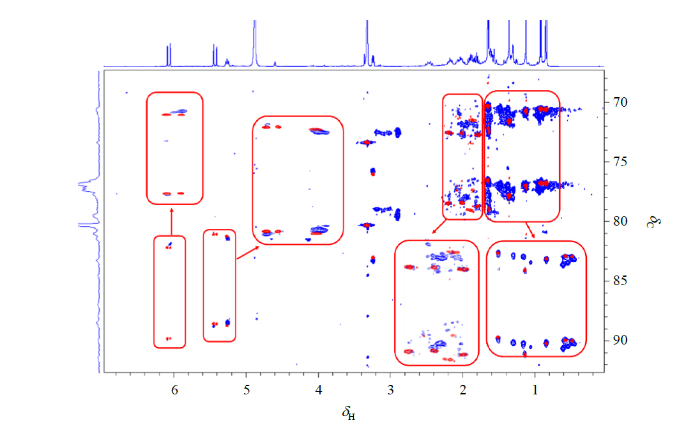
图2
Sinulariol Z在各向同性相(MeOD-d4,红色等高线)和各向异性相(40 mg/mL AAKLVFF LCs,蓝色等高线)中的perfect 1JCH -resolved HSQC覆盖谱图
Fig. 2
Overlaid perfect 1JCH -resolved HSQC spectra of sinulariol Z in the isotropic phase (MeOD-d4, red contours) and in the anisotropic phase (40 mg/mL AAKLVFF LCs, blue contours)
表1 Sinulariol Z的实验和理论RDC值
Table 1
| Position | Sinulariol Z | |
|---|---|---|
| Exp. 1DCH/Hz | Calc. 1DCH/Hz (S*S*S*R*) | |
| C2-H2 | 8.104 | 8.74 |
| C3-H3 | 10.54 | 9.49 |
| C6-H6a | -16.524 | -10.51 |
| C9-H9a | 0.79 | 3.24 |
| C9-H9b | -1.346 | 1.96 |
| C11-H11 | 9.572 | 10.15 |
| C15-H15 | 1.614 | 3.53 |
| C16-H16 | -0.92 | 1.63 |
| C17-H17 | 0.572 | 0.34 |
| C18-H18 | -3.562 | -1.93 |
| C19-H19 | 2.486 | 2.93 |
| C20-H20 | -2.15 | -2.08 |
对化合物sinulariol Z所有可能存在的非对映异构体进行构象搜索、DFT优化以及振动频率分析,并根据玻尔兹曼分布理论和相对吉布斯自由能计算出各构象的玻尔兹曼分布比例(见附件图S1、表S1和表S2). 通过Mspin软件的单张量近似法分析,最终得到sinulariol Z的理论推测RDC值(表1).
将sinulariol Z的实验RDC值与理论推测RDC值进行Q因子拟合,拟合结果如图3所示,图中1~8组代表的相对构型分别为(1S*、4S*、11S*、12R*),(1R*、4S*、11S*、12R*),(1S*、4S*、11S*、12S*),(1S*、4S*、11R*、12R*),(1R*、4R*、11S*、12R*),(1S*、4R*、11S*、12R*),(1S*、4R*、11R*、12R*)和(1S*、4R*、11S*、12S*),其中Q因子值最小的相应立体构型,可认为是sinulariol Z的正确相对构型,即(1S*、4S*、11S*、12R*).
图3
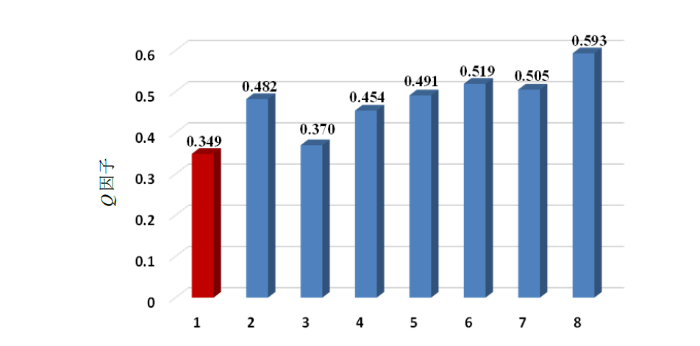
图3
Sinulariol Z所有相对构型的Q因子(第1组为Q因子最小值,即sinulariol Z的正确构型)
Fig. 3
The Q factors for relative configurations of sinulariol Z (Group 1 has the minimum Q factor, which is the correct configuration of sinulariol Z)
2.3 单晶X射线衍射法佐证sinulariol Z的立体结构
为了验证RDC所指向的sinulariol Z构型的正确性,我们在乙醇/水体系中成功培养出了sinulariol Z的单晶(图4).通过低温铜靶单晶衍射确定的sinulariol Z晶体结构(CCDC 2130603)准确确定了sinulariol Z的绝对构型[Flack参数(Flack parameter):0.29(18)]为(1S、4S、11S、12R).由此可知,RDC能够准确确定该西松烷二萜的立体构型.
图4
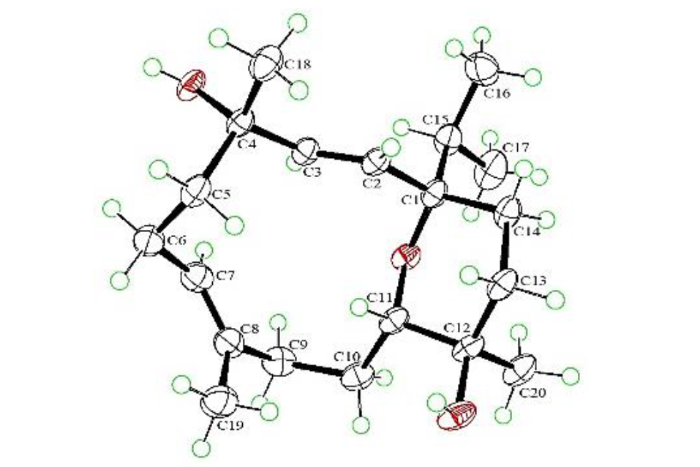
图4
化合物sinulariol Z的单晶结构
Fig. 4
Perspective ORTEP drawing of the X-ray structure of sinulariol Z
3 结论
RDC能够提供化合物整体分子的构型信息,在构建分子的立体空间结构方面已经成为一种有效的方法,不仅能够用于研究生物大分子的结构和功能,而且在研究天然产物分子的立体构型方面也显现出了独特的优势.然而RDC在西松烷二萜类化合物立体构型确定方面的应用较少,尚缺乏非常充足且可观的实验数据.因此,本文对西松烷二萜类化合物sinulariol Z首次采用RDC确定其立体构型,并结合单晶X衍射方法对该化合物的立体构型进行了再次确证,进一步验证了RDC在该类化合物的立体构型确定方面的可行性.
在西松烷二萜类化合物立体构型研究中,RDC的实验值和理论预测值的拟合能够达到非常好的效果,如化合物sarcomililate A的Q因子能够达到0.08[24].然而,有时候RDC在这类化合物的立体构型确定方面展现的效果并不理想,如本文所测定的化合物sinulariol Z,指向正确相对构型的Q因子(0.349)且与指向错误相对构型的Q因子(0.370)相差较小,但是单晶X衍射实验结果证明该拟合结果是正确的,能够准确确定化合物sinulariol Z的立体构型.由于RDC反映的是化合物化学键的全局性结构信息,其结果与溶液中的构象密切相关,因此推测导致拟合结果不理想可能是由于该类化合物偏柔性,构象抓取困难,致使构象抓取范围不够准确[14].
利益冲突
无
附件材料附录(可在期刊官网论文网页版获取)
图S1 Sinulariol Z各非对映异构体(玻尔兹曼比例大于1%)的结构信息
图S2 Sinulariol Z的1H NMR谱图
图S3 Sinulariol Z的13C NMR谱图
表S1 Sinulariol Z各非对映异构体的优势构象的笛卡尔坐标
表S2 Sinulariol Z各非对映异构体构象的玻尔兹曼比例
参考文献
Several stereogenic problems regarding structural elucidation of marine natural products
[J].
海洋天然产物结构解析若干立体化学问题
[J].
Marine natural products
[J].DOI:10.1039/c005001f PMID:21152619 [本文引用: 1]
Discovery of benthol A and its challenging stereochemical assignment: opening up a new window for skeletal diversity of super-carbon-chain compounds
[J].
Dumulmycin, an antitubercular bicyclic macrolide from a riverine sediment-derived streptomyces sp.
[J].
Racemic bisindole alkaloids: structure, bioactivity, and computational study
[J].
Discovery and structural characterization of impurities in the synthesis of darolutamide intermediate
[J].
达罗他胺中间体合成中杂质的发现和结构表征
[J].
DOI:10.11938/cjmr20233078
[本文引用: 1]

达罗他胺是治疗前列腺癌的重要药物,在进行其合成工艺研究时,在第一步Suzuki偶联和第二步水解脱保护反应中,发现并纯化得到3个杂质A、B和C,其中杂质A和B来自第一步反应,杂质C来自第二步反应. 通过高分辨质谱(HRMS)、核磁共振氢谱(<sup>1</sup>H NMR)和核磁共振碳谱(<sup>13</sup>C NMR)表征方法,确定了杂质A和B的结构,分别为初始反应原料化合物2的脱硼酸频哪醇酯产物和初始反应原料化合物1的双偶联产物;借助HRMS、<sup>1</sup>H NMR、<sup>13</sup>C NMR、<sup>1</sup>H-<sup>1</sup>H COSY、<sup>1</sup>H-<sup>13</sup>C HSQC、<sup>1</sup>H-<sup>13</sup>C HMBC和<sup>1</sup>H-<sup>1</sup>H NOESY方法确定了中间体化合物3和杂质C的准确结构,对其形成机理和规避方法也进行了讨论分析.
Structural elucidation of hybutimibe
[J].
海博麦布结构确证
[J].
DOI:10.11938/cjmr20233065
[本文引用: 1]
本文采用紫外吸收光谱、红外吸收光谱、质谱、核磁共振波谱(包含<sup>1</sup>H NMR、<sup>13</sup>C NMR、DEPT、<sup>1</sup>H-<sup>1</sup>H COSY、<sup>1</sup>H-<sup>1</sup>H NOESY、<sup>1</sup>H-<sup>13</sup>C HSQC和<sup>1</sup>H-<sup>13</sup>C HMBC)以及单晶衍射等方法对海博麦布进行结构分析,对其所有的<sup>1</sup>H NMR和<sup>13</sup>C NMR谱信号进行了归属,还通过差示扫描量热法、热重分析及粉末X-射线衍射分析对海博麦布晶型进行研究.
Application of quantum chemical calculation of nuclear magnetic resonance parameters in the structure elucidation of natural products
[J].
量子化学计算核磁共振参数在天然产物结构鉴定中的应用
[J].
DOI:10.11938/cjmr20182682
[本文引用: 1]
在过去的十多年中,伴随着量子化学理论与计算机硬件、软件的不断发展,量子化学计算核磁共振参数(quantum chemical calculation of nuclear magnetic resonance parameters,qcc-NMR)的方法也日趋成熟,这些方法往往在较小的计算成本下就可以获得比较理想的计算精度,且对于NMR参数计算结果的分析也从最初的简单统计学方法逐渐发展为基于更为复杂的统计学原理或人工神经网络的方法,这些进展都促使qcc-NMR这一工具在天然产物研究中得到了越来越广泛的应用,从而对传统的NMR技术、质谱,以及各种光谱技术做出了重要补充.本文对qcc-NMR在天然产物结构鉴定中的应用进行了综述,并对近年来的一些应用实例进行了较为详细的分析.
Beyond DP4: an improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts
[J].
DOI:10.1021/acs.joc.5b02396
PMID:26580165
[本文引用: 1]
The DP4 probability is one of the most sophisticated and popular approaches for the stereochemical assignment of organic molecules using GIAO NMR chemical shift calculations when only one set of experimental data is available. In order to improve the performance of the method, we have developed a modified probability (DP4+), whose main differences from the original DP4 are the inclusion of unscaled data and the use of higher levels of theory for the NMR calculation procedure. With these modifications, a significant improvement in the overall performance was achieved, providing accurate and confident results in establishing the stereochemistry of 48 challenging isomeric compounds.
Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability
[J].
DOI:10.1021/ja105035r
PMID:20795713
[本文引用: 1]
GIAO NMR shift calculation has been applied to the challenging task of reliably assigning stereochemistry with quantifiable confidence when only one set of experimental data are available. We have compared several approaches for assigning a probability to each candidate structure and have tested the ability of these methods to distinguish up to 64 possible diastereoisomers of 117 different molecules, using NMR shifts obtained in rapid and computationally inexpensive single-point calculations on molecular mechanics geometries without time-consuming ab initio geometry optimization. We show that a probability analysis based on the errors in each (13)C or (1)H shift is significantly more successful at making correct assignments with high confidence than are probabilities based on the correlation coefficient and mean absolute error parameters. Our new probability measure, which we have termed DP4, complements the probabilities obtained from our previously developed CP3 parameter, which applies to the case of assigning a pair of diastereoisomers when one has both experimental data sets. We illustrate the application of DP4 to assigning the stereochemistry or structure of 21 natural products that were originally misassigned in the literature or that required extensive synthesis of diastereoisomers to establish their stereochemistry.
DP4-AI automated NMR data analysis: straight from spectrometer to structure
[J].
DOI:10.1039/d0sc00442a
PMID:34122893
[本文引用: 1]
A robust system for automatic processing and assignment of raw C and H NMR data DP4-AI has been developed and integrated into our computational organic molecule structure elucidation workflow. Starting from a molecular structure with undefined stereochemistry or other structural uncertainty, this system allows for completely automated structure elucidation. Methods for NMR peak picking using objective model selection and algorithms for matching the calculated C and H NMR shifts to peaks in noisy experimental NMR data were developed. DP4-AI achieved a 60-fold increase in processing speed, and near-elimination of the need for scientist time, when rigorously evaluated using a challenging test set of molecules. DP4-AI represents a leap forward in NMR structure elucidation and a step-change in the functionality of DP4. It enables high-throughput analyses of databases and large sets of molecules, which were previously impossible, and paves the way for the discovery of new structural information through machine learning. This new functionality has been coupled with an intuitive GUI and is available as open-source software at https://github.com/KristapsE/DP4-AI.This journal is © The Royal Society of Chemistry.
Stereochemical investigation of flexible macrocyclic cembranes depending on residual dipolar couplings method
[J].
Structural elucidation of organic molecule from residual dipolar couplings
[J].
核磁共振残留偶极耦合参数在有机分子结构鉴定中的应用
[J].
Residual dipolar couplings and their applications in determination of protein structures
[J].
残留偶极耦合及其在蛋白质结构研究中的应用
[J].近年来溶液中残留偶极耦合常数被用来获取生物大分子化学键之间相对取向等长程构象约束条件,用于计算或优化蛋白质及其复合物的三维空间结构. 介绍了用异核多维NMR技术测量残留偶极耦合常数的方法,及其在蛋白质结构计算中的一些应用:优化蛋白质溶液结构,评价蛋白质结构质量,确定蛋白质结构域取向,获取有关配体的构象和取向的信息,在缺乏NOE数据时构建蛋白质结构等.
Residual dipolar couplings in structure determination of natural products
[J].
Dysoxylactam A: A macrocyclolipopeptide reverses P-glycoprotein-mediated multidrug resistance in cancer cells
[J].
Investigation of dynamic structure of protein encountering complex with paramagnetic NMR
[J].
顺磁核磁共振技术研究蛋白质遭遇复合物的动态结构
[J].
DOI:10.11938/cjmr20223035
[本文引用: 1]
蛋白质依靠短程相互作用识别配体蛋白进而行使生物学功能,其相互作用界面仅占据蛋白质总表面积的一部分.因此,蛋白质与配体蛋白需要形成一系列遭遇复合物系综结构来减少构象搜索空间以加快结合速度.由于遭遇复合物在溶液体系中存在时间短、丰度低,因而很难被传统结构生物学技术捕捉到.本文选用组氨酸磷酸载体蛋白(HPr)和酶II(EIIA<sup>Glc</sup>)复合物为研究体系,采用顺磁弛豫增强(Paramagnetic Relaxation Enhancement,PRE)技术对遭遇复合物的系综结构及动力学性质进行表征,并用分子动力学模拟方法对实验结果进行验证,发现HPr在溶液体系中首先与EIIA<sup>Glc</sup>在3个方向上形成遭遇复合物,进而促进特异性复合物的形成.该方法不仅能够在溶液体系中观察遭遇复合物系综结构,还有望应用于生物大分子领域,揭示蛋白质在复杂生理网络中的相互作用机制及动力学行为.
Measurement of residual dipolar couplings of organic molecules in multiple solvent systems using a liquid-crystalline-based medium
[J].
RDC-enhanced NMR spectroscopy in structure elucidation of sucro-neolambertellin
[J].DOI:10.1002/anie.200705037 PMID:18236483 [本文引用: 1]
Sarcomililate A, an unusual diterpenoid with tricyclo[11.3.0.02,16]hexadecane carbon skeleton, and its potential biogenetic precursors from the Hainan soft coral Sarcophyton mililatensis
[J].
α-Methylene-γ-lactone-bearing cembranoid diterpenes from the south China sea soft coral Lobophytum sp.
[J].
中国南海豆荚软珊瑚Lobophytum sp.中西松烷二萜类化学成分研究
[J].
Cembrane diterpenes chemistry and biological properties
[J].
Cembranoids from the soft coral sinularia rigida with antifouling activities
[J].
Perfect 1JCH -resolved HSQC: Efficient measurement of one-bond proton-carbon coupling constants along the indirect dimension
[J].
A program for the use of residual dipolar couplings for structure elucidation of small molecules
[J].
A self-assembled oligopeptide as a versatile NMR alignment medium for the measurement of residual dipolar couplings in methanol
[J].
DOI:10.1002/anie.201705123
PMID:28834640
[本文引用: 1]
Residual dipolar coupling (RDC) is a powerful structural parameter for the determination of the constitution, conformation, and configuration of organic molecules. Herein, we report the first liquid crystal-based orienting medium that is compatible with MeOH, thus enabling RDC acquisitions of a wide range of intermediate to polar organic molecules. The liquid crystals were produced from self-assembled oligopeptide nanotubes (AAKLVFF), which are stable at very low concentrations. The presented alignment medium is highly homogeneous, and the size of RDCs can be scaled with the concentration of the peptide. To assess the accuracy of the RDC measurement by employing this new medium, seven bioactive natural products from different classes were chosen and analyzed. The straightforward preparation of the anisotropic alignment sample will offer a versatile and robust protocol for the routine RDC measurement of natural products.© 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Anti-inflammatory cembrane-type diterpenoids and prostaglandins from soft coral lobophytum sarcophytoides
[J].
Computer-assisted 3D structure elucidation (CASE-3D) of natural products combining isotropic and anisotropic NMR parameters
[J].
NMR-based configurational assignments of natural products: how floating chirality distance geometry calculations simplify gambling with 2N-1 diastereomers
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}