


引言
前列腺癌(Prostate cancer)是发生在前列腺的上皮恶性肿瘤,是男性泌尿生殖系统最常见的恶性肿瘤. 该疾病在老年男性中发病率极高,现呈现低龄化且全球发病率持续上升,高居男性恶性肿瘤发病率第二位[1,2]. 达罗他胺(Darolutamide,NUBEQA,7)是一种非甾体类雄激素受体(Androgen receptor,AR)抑制剂,具有独特于其它雄激素受体抑制剂的化学结构,能够竞争性的抑制雄激素与AR结合、AR核转运和AR介导的转录,展现出很强的拮抗活性,从而抑制前列腺癌细胞的增殖,缩小肿瘤体积. 该药由芬兰Orion公司与德国Bayer公司联合开发,目前已经在美国、欧盟、日本、中国等全球70多个国家和地区获得批准,广泛用于治疗非转移性去势抵抗性前列腺癌(Non-metastatic castration-resistant prostate cancer, nmCRPC)患者[3⇓-5].
图1

图1
达罗他胺(7)的原研合成路线. THF:四氢呋喃;PPh3:三苯基膦;DIAD:偶氮二碳酸二异丙酯;DIPEA:N,N-二异丙基乙胺;HOBt:1-羟基苯并三唑;EDCI:1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐
Fig. 1
Original synthesis route of darolutamide (7). THF: tetrahydrofuran; PPh3: triphenylphosphine; DIAD: diisopropyl azodicarboxylate; DIPEA: N,N-diisopropylethylamine; HOBt: 1-hydroxybenzotriazole; EDCI: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
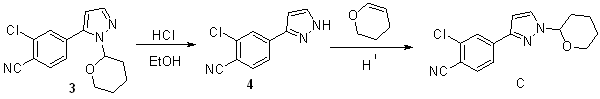
本文采用以上合成路线对达罗他胺的合成工艺进行研究. 在合成中间体2-氯-4(1-(四氢-2H-吡喃-2-基)-1H-吡唑-5-基)苯甲腈(化合物3)时,通过中控高效液相色谱(HPLC)检测,在反应液中发现两个含量较高的未知杂质(分别标记为杂质A和B,HPLC含量分别为3.90%和0.61%,见附件材料图S1).在合成2-氯-4(1H-吡唑-3-基)苯甲腈(化合物4)时,在反应液中发现一个含量较高的杂质(标记为杂质C,HPLC含量1.23%,见附件材料图S2),且随着反应时间的延长,杂质C的含量增加. 经文献检索,这3个杂质的结构及形成过程在达罗他胺的工艺研究中未见有相关报道. 另一方面,在药物合成工艺研究中,除了对反应条件和后处理方式进行系列优化外,药物杂质的结构确定也对工艺优化具有非常重要的指导意义,例如可以为杂质的形成、传递、去除和限度制定等提供直接的参考价值. 基于此,本文以4-溴-2-氯苯甲腈(化合物1)为原料,首先与1-(2-四氢吡喃基)吡唑-5-硼酸频哪醇酯(化合物2)在钯催化下发生Suzuki偶联,随后在盐酸-乙醇溶液中脱保护基四氢吡喃(THP),得到化合物4(图2).为进一步确证杂质结构,我们在两步反应中收集并纯化得到三个杂质A、B和C,HPLC检测纯度均在95%以上. 对于杂质A和B,因其结构与产物3相差较大,利用HRMS和一维NMR(1H NMR和13C NMR)即可确定其结构;而杂质C为化合物3的同分异构体,综合运用HRMS、一维和二维NMR(1H-1H COSY、1H-13C HSQC、1H-13C HMBC和1H-1H NOESY)对其进行结构解析,同时对产生异构体C的反应历程也进行了推测. 研究结果对达罗他胺的合成工艺研究和原料药的质量控制具有重要的指导作用.
图2

1 实验部分
1.1 仪器与试剂
实验所用起始物4-溴-2-氯苯腈(化合物1)购于浙江省平湖市艾柏化工有限公司;1-(2-四氢吡喃基)吡唑-5-硼酸频哪醇酯(化合物2)购于上海韶远试剂有限公司;双三苯基膦二氯化钯(Pd(Ph3)2Cl2)购于上海凌凯医药科技有限公司;氘代氯仿(CDCl3)和氘代二甲基亚砜(DMSO-d6)(含0.03%的四甲基硅烷(TMS))购于北京伊诺凯科技有限公司;薄层板GF254、层析硅胶(200~300目)、碳酸钠、三乙胺、盐酸、四氢呋喃(THF)、乙腈、乙醇、正己烷和乙酸乙酯等均为国产市售试剂. 所有试剂均未进一步纯化.
一维(1H NMR和13C NMR)和二维(1H-1H COSY、1H-13C HSQC、1H-13C HMBC和1H-1H NOESY)NMR数据采集在Bruker AVANCE III 400/600 MHz核磁共振波谱仪上进行.1H NMR的工作频率为400 MHz或600 MHz,13C NMR的工作频率为101 MHz或151 MHz,实验温度为298.2 K,均采用标准脉冲程序. 高分辨质谱(HRMS)采用Agilent 1260 Infinity/6530 Q-TOF系统,配备电喷雾离子源(ESI).
1.2 化合物3、杂质A和B的合成
化合物3参照文献报道方法制备[7⇓-9],合成路线如图2所示,具体方法如下:将化合物1(8.00 g, 36.96 mmol)、化合物2(12.95 g,46.57 mmol)、碳酸钠(9.40 g,88.70 mmol)、24 mL水和Pd(PPh3)2Cl2(0.13 g,0.19 mmol)依次加入到80 mL THF中,氮气置换3次,置于预先加热至45 ℃的恒温油浴中搅拌反应3 h. 经薄层色谱(TLC,V正己烷:V乙酸乙酯 = 10:1)检测至化合物1反应完全后,将反应液降温至30 ℃. 减压旋转蒸发浓缩去除有机相,向残留液中加入96 mL水,在30 ℃下搅拌30 min后过滤,滤饼用无水乙醇(64 mL)打浆,并在30 ℃下搅拌1 h,降温至-10 ℃继续搅拌1 h后过滤. 滤饼于50 ℃下真空干燥12 h,得化合物3(7.60 g,收率71.5%).将上述抽滤后的滤液浓缩,残留液经柱层析梯度洗脱分离(洗脱剂为正己烷和乙酸乙酯的混合液,v/v,50:1、20:1、15:1、8:1),分别得杂质B(白色固体,0.10 g)和杂质A(无色油状,0.22 g).杂质A和B的1H NMR和13C NMR数据(谱图见附件材料图S3和S4)和HRMS数据结果如下:
杂质A:1H NMR (400 MHz, CDCl3):δH 7.57 (d, J = 2.5 Hz, 1H),7.52 (s, 1H),6.27 (t, J = 2.2 Hz, 1H),5.36 (dd, J = 9.7, 2.7 Hz, 1H),4.04~4.00 (m, 1H),3.69-3.63 (m, 1H),2.12~1.99 (m, 3H),1.68~1.55 (m, 3H). 13C NMR (101 MHz,CDCl3):δC 139.54,127.56,105.97,87.52,67.78,30.50,24.96,22.48. HRMS (ESI) m/z:[M+H]+ C8H13N2O+,理论值:153.102 2,实测值:153.102 7.
杂质B:1H NMR (400 MHz, CDCl3):δH 7.96~7.85 (m, 2H),7.78~7.71 (m, 1H),7.69~7.63 (m, 2H),6.61 (dd, J = 20.0, 1.6 Hz, 1H),6.46 (dd, J = 6.8, 1.6 Hz, 1H),5.18~5.12 (m, 2H),4.10~3.94 (m, 2H),3.56~3.48 (m, 2H),2.59~2.55 (m, 2H),2.12~1.86 (m, 3H),1.74~1.52 (m, 5H), 1.27~1.25 (m, 1H),0.88 (t, J = 6.4 Hz, 1H). 13C NMR (101 MHz, CDCl3):δC 141.79,139.72, 139.64,139.54, 138.98,138.86, 134.91,134.89,134.50,134.43, 133.89,133.75,131.42,131.36, 130.96,130.92, 129.12,117.39,117.25, 112.74,112.60, 109.13,109.08,109.03,108.97,107.81,107.76, 107.69,107.65,84.60, 84.57,67.62, 67.59,67.55,67.37,29.51, 29.33,29.30, 24.79,24.71, 22.82,22.58. HRMS (ESI) m/z:[M+K]+ C23H25KN5O2+,理论值:442.164 0,实测值:442.164 6.
1.3 化合物4和杂质C的合成
化合物4参照文献报道方法制备[7⇓-9],合成路线如图2所示,具体方法如下:将化合物3(10.00 g,34.83 mmol)溶于40 mL乙醇中,在25 ℃下缓慢滴加10 wt%的盐酸水溶液(2.54 g,含HCl 6.97 mmol),滴加完毕后,置于预先加热至35 ℃的恒温油浴中搅拌反应1 h. TLC(V正己烷:V乙酸乙酯 = 10:1)检测至化合物3反应完全. 随后,向反应液中滴加10 wt%的NaOH水溶液调节pH≈10,降温至0 ℃后继续搅拌1 h后过滤,滤饼用20 mL水淋洗后,于60 ℃下真空干燥12 h,得化合物4(6.67 g,收率94.1%),其1H NMR和13C NMR数据(谱图见附件材料图S5)和高HRMS结果如下:
化合物4:1H NMR (400 MHz, DMSO-d6):δH 13.27 (s, 1H),8.14 (s, 1H),7.98 (s, 2H),7.88 (d, J = 1.6 Hz, 1H),6.98 (d, J = 1.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6):δC 147.85,140.53,136.33,135.41,131.18,126.21,124.60,116.71,110.31,104.02. HRMS (ESI) m/z:[M+H]+ C10H7ClN3+,理论值:204.032 3,实测值:204.033 0.
如前所述,化合物4在制备过程时,HPLC检测反应液中其含量为1.23%(附件材料图S2).而在获得上述固体化合物4后,对其再次进行HPLC检测,发现固体中仍然混有杂质C,含量为1.50%,稍高于反应液中的含量(附件材料图S6).为了能更快的富集得到该杂质以便对其进行结构确证,我们对化合物4的合成条件进行了适当的调整,提高了反应温度并适当延长了反应时间,具体方法如下:将化合物3(10.00 g,34.83 mmol)溶于40 mL乙醇中,在25 ℃下滴加10 wt%的盐酸水溶液(2.54 g,含HCl 6.97 mmol),滴加完毕后升温至60 ℃下搅拌反应6 h. 随后将反应液冷却至室温,向其滴加10 wt%的NaOH水溶液调节pH10,降温至0 ℃继续搅拌1 h后过滤,滤饼用20 mL水淋洗,收集滤饼即为化合物4的粗品. 粗品HPLC检测,杂质C的含量增加至17.34%(附件材料图S7).柱层析梯度洗脱分离(洗脱剂为正己烷和乙酸乙酯的混合液,v/v,50:1~10:1)后,得到杂质C(0.83 g).
2 结果与讨论
2.1 杂质A和B的分析
在第1步Suzuki偶联反应的中控HPLC分析发现,反应液中主要含有少量未反应完的原料化合物1和化合物2、产物即化合物3、三苯基氧磷(来自于钯催化剂)、杂质A和B,以及少量脱THP产物即化合物4(即第2步产物);而按实验部分1.2节所述的操作,得到经真空干燥后的固体化合物3中仍含有其它组分,如原料化合物1、三苯基氧磷、杂质A和B,以及化合物4,具体的HPLC含量如表1所示(谱图见附件材料图S1和S8).经HRMS、1H NMR和13C NMR表征,杂质A为原料化合物2的脱硼酸频哪醇酯产物,而杂质B则为原料化合物1的双偶联产物. 这两个杂质的含量与反应中原料化合物2的用量、钯催化剂、反应温度、碱的强弱均有一定的关系. 反应结束后,产物3主要通过乙醇打浆的方式纯化,得到的固体中,化合物3的含量由92.75%提高到98.46%,杂质A和B的含量则由3.90%和0.61%分别下降至0.04%和0.14%.由此可知,无水乙醇打浆处理的方式对产物3的纯化是比较成功的,大部分的杂质A均留在母液中,且其在化合物3中的少量残留对后续反应基本没有影响. 而杂质B因含有类似化合物3的结构单元,它的残留会参与后续反应,但因其只有0.14%的含量,在后续中间体合成中又有其它的纯化步骤,所以对终产品影响不大.如杂质B含量较大,需考虑对终产品质量的影响.
表1 Suzuki偶联反应中各组分HPLC含量分析
Table 1
| 名称 | 化合物1 | 化合物2 | 化合物3 | 三苯基氧磷 | 杂质A | 杂质B | 化合物4 |
|---|---|---|---|---|---|---|---|
| 反应液中含量 | 0.25% | 1.10% | 92.75% | 0.52% | 3.90% | 0.61% | 0.67% |
| 固体中含量 | 0.35% | 0 | 98.46% | 0.07% | 0.04% | 0.14% | 0.85% |
2.2 化合物3和杂质C的HRMS图谱分析
化合物3(C15H14ClN3O)和杂质C(C15H14ClN3O)的HRMS图见附件材料图S9. 图S9(a)中信号最强峰[m/z 204.031 3]为准分子离子峰[M-THP]+,图S9(b)中信号最强峰[m/z 310.073 0]为准分子离子峰[M+Na]+,分别与两个化合物的理论值([M-THP]+:m/z 204.032 3和[M+Na]+:m/z 310.071 8)接近. 因化合物3和杂质C是异构体,仅从质谱图无法确定二者的准确结构.
2.3 化合物3和杂质C的NMR数据分析
化合物3和杂质C的化学结构及相关碳原子的编号如图2所示.
2.3.1 化合物3和杂质C的1H NMR和13C NMR
化合物3和杂质C的1H NMR谱图见附件材料图S10所示. 两个化合物的1H NMR谱除高场吡喃环部分有些许差异外,其它部分比较相似,均显示有13组质子信号. 低场区δH 7.58~7.79为苯环的三个质子信号和吡唑环上的一个质子信号;δH 6.46归属为H-9;δH 5.15归属为H-11;δH 4.17、δH 3.64归属为H-15,δH 1.62~2.59为H-12、H-13和H-14. 可能由于吡喃环在空间距离上与苯环靠的更近,使得化合物3中H-12的两组氢的化学位移相差较大,分别为δH 2.59和δH 1.90,而杂质C中H-12的化学位移则为δH 2.19和δH 2.13. 化合物3和杂质C的1H-1H NOESY结果也证实这个推测(见附件材料图S11和S12).其中,化合物3中的H-6与H-11,H-7与H-11和H-15相关;而在杂质C中,则可以看到H-10与H-11和H-12相关. 然而,这种差异还不足以解析两个化合物的准确结构,需要借助其他二维谱进一步确认.
化合物3和杂质C的13C NMR谱图见附件材料图S13所示. 两个化合物均显示有15组碳原子信号. 主要的不同点是图S13(a)的δC 141.1与图S13(b)的δC 148.5,其它的信号都很相近. 图S13(a)中,δC 108.0~141.1共有10个碳,归属于苯环、吡唑环和与苯环相连的氰基碳;δC 84.6归属于吡喃环上的C-11;δC 67.6归属于吡喃环上的C-15;δC 22.8~29.5上的3个碳归属于C-12、C-13和C-14,具体的精确归属需要进一步借助1H-1H COSY、1H-13C HSQC和1H-13C HMBC.
2.3.2 化合物3和杂质C的二维NMR
图3
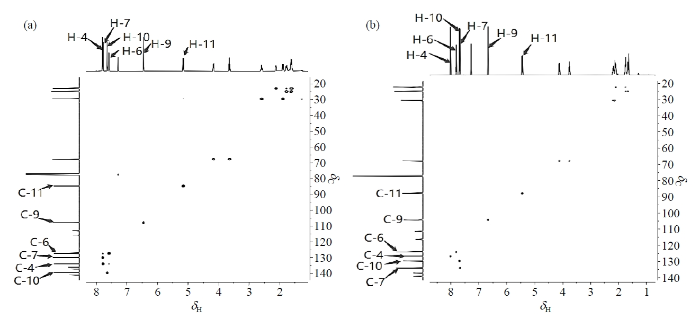
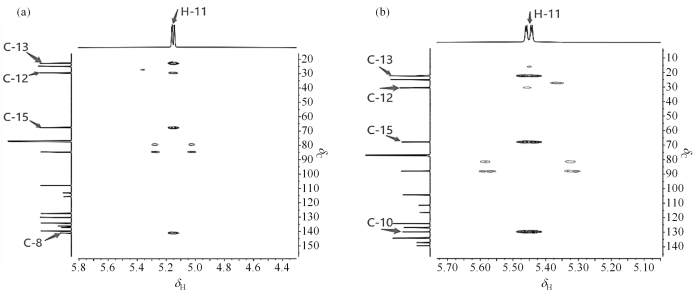
图4

图5

以化合物3为例,在图3(a)的1H-13C HSQC谱图中可以看到δC 108.0和δC 84.6分别与前面一维NMR确定的H-9和H-11相关,归属为C-9和C-11. 在图4(a)的1H-1H COSY谱图中,从H-9的δH 6.46可以找到与其相关的δH 7.64,归属为H-10. 由1H-13C HSQC归属与H-10相关的C-10即δC 139.7. 通过图5(a)的1H-13C HMBC的图,H-10与C-8和C-9相关,找到δC 141.1,归属为C-8. H-9与C-5、C-8和C-10相关,确定C-5即δC 136.2. H-11与C-8、C-12、C-13和C-15相关,找到δC 29.5、δC 22.8和δC 67.6,分别归属为C-12、C-13和C-15. H-14与C-12、C-13和C-15相关,找到δH 1.62与δH 1.78,归属为H-14. 通过1H-1H COSY可看出δH 7.59与δH 7.78相关,再通过1H-13C HMBC找到与C-8相关的只有H-6,所以,δH 7.59与δH 7.78分别归属H-6和H-7,进一步通过1H-13C HSQC分别找到δC 127.4和δC 134.1,归属为C-6和C-7. H-7相关的C-1、C-3和C-5,其中C-5对应的前面已归属,因C-3与氯原子相连,去屏蔽作用更强,所以δC 137.2归属为C-3,δC 115.7归属为C-1. 至此,最后一个氢δH 7.79归属为H-4,根据1H-13C HSQC 找到δC 130.2,归属为C-4. 通过1H-13C HMBC,找到与H-4相关的C-2、C-6和C-8,其中C-6和C-8前面已经归属,所以δC 113.0归属为C-2. 综上,借助二维NMR准确地归属了化合物3的所有NMR数据.
表2 化合物3的NMR数据归属
Table 2 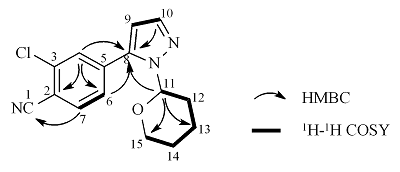
| No. | 1H NMR | 13C NMR | HMBC(1H→13C) | 1H-1H COSY |
|---|---|---|---|---|
| δH | δC | |||
| 1 | N/A | 115.7 | N/A | N/A |
| 2 | N/A | 113.0 | N/A | N/A |
| 3 | N/A | 137.2 | N/A | N/A |
| 4 | 7.79 (1H, d, J=1.4 Hz) | 130.2 | C-2, 6, 8 | N/A |
| 5 | N/A | 136.2 | N/A | N/A |
| 6 | 7.59 (1H, dd, J=8.0, 1.5 Hz) | 127.4 | C-2, 3, 4, 7, 8 | H-7 |
| 7 | 7.78 (1H, d, J=8.1 Hz) | 134.1 | C-1, 3, 5 | H-6 |
| 8 | N/A | 141.1 | N/A | N/A |
| 9 | 6.46 (1H, d, J=1.8 Hz) | 108.0 | C-5, 8, 10, 11 | H-10 |
| 10 | 7.64 (1H, d, J=1.6 Hz) | 139.7 | C-8, 9 | H-9 |
| 11 | 5.15 (1H, dd, J=10.0, 2.4 Hz) | 84.6 | C-8, 12, 13, 15 | H-12 |
| 12 | 1.90 (1H, m), 2.59 (1H, m) | 29.5 | C-11, 13, 14 | H-11, 13 |
| 13 | 1.62 (1H, overlap), 2.12 (1H, m) | 22.8 | C-11, 12, 14, 15 | H-12, 14 |
| 14 | 1.62 (1H, overlap), 1.78 (1H, m) | 24.8 | C-12, 13, 15 | H-13, 15 |
| 15 | 3.64 (1H, m), 4.17 (1H, m) | 67.6 | C-11, 13, 14 | H-14 |
表3 杂质C的NMR数据归属
Table 3 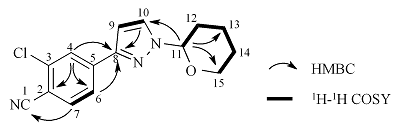
| No. | 1H NMR | 13C NMR | HMBC(1H→13C) | 1H-1H COSY |
|---|---|---|---|---|
| δH | δC | |||
| 1 | N/A | 116.3 | N/A | N/A |
| 2 | N/A | 111.6 | N/A | N/A |
| 3 | N/A | 137.1 | N/A | N/A |
| 4 | 8.01 (1H, d, J=1.4 Hz) | 126.7 | C-2, 3, 6, 8 | N/A |
| 5 | N/A | 139.3 | N/A | N/A |
| 6 | 7.81 (1H, dd, J=8.1, 1.5 Hz) | 124.1 | C-2, 4, 8 | H-7 |
| 7 | 7.68 (1H, d, J=8.1 Hz) | 134.1 | C-1, 3, 5 | H-6 |
| 8 | N/A | 148.5 | N/A | N/A |
| 9 | 6.67 (1H, d, J=2.5 Hz) | 104.2 | C-8, 10 | H-10 |
| 10 | 7.70 (1H, d, J=2.5 Hz) | 129.7 | C-8, 9 | H-9 |
| 11 | 5.45 (1H, dd, J=9.6, 2.6 Hz) | 88.0 | C-10, 12, 13, 15 | H-12 |
| 12 | 2.13 (1H, m), 2.19 (1H, m) | 30.6 | C-11, 13, 14 | H-11, 13 |
| 13 | 1.75 (1H, overlap), 2.09 (1H, m) | 22.3 | C-11, 12, 14, 15 | H-12, 14 |
| 14 | 1.67 (1H, m), 1.75 (1H, overlap) | 24.9 | C-12, 13, 15 | H-13, 15 |
| 15 | 3.76 (1H, m), 4.11 (1H, m) | 68.0 | C-11, 13, 14 | H-14 |
图6
2.4 杂质C形成机理及影响因素
从杂质C的结构来看,形成机理如图7所示. 推测为在酸性条件下,化合物3脱保护生成化合物4和THP;而化合物4在酸性条件下可重新与THP反应生成杂质C. 该反应历程表明杂质C比化合物3在酸性条件下的稳定性更好,并且随着反应温度的提高和反应时间的延长,杂质C含量会进一步增加.
图7
为了验证这个结论,作者研究了反应温度和反应时间对反应产物和杂质C的影响,结果如表4所示. 当反应温度为20 ℃时,该反应进行1 h(编号1),产物化合物4的含量仅为33.25%;延长反应时间到5 h,则基本反应完全,进一步延长至22 h,产物4的含量基本没变化,但杂质C的含量由0.47%增加到1.20%(编号2~3).升高反应温度到30 ℃或40 ℃时,该反应在1 h内即可反应完全,但杂质C的含量均比20 ℃反应时要高,并且随着反应时间的延长,杂质C含量也增多(编号4~7).当反应温度继续升高到50 ℃或60 ℃时,反应1 h后的结果均显示产物化合物4的含量下降,而杂质C的含量则显著上升,温度越高该趋势越明显(编号6、8和9).因此,对于第二步脱保护基THP,反应温度选择20~30 ℃,反应时间控制在5 h以内为最佳,此时产物化合物4的含量高,杂质C的含量较低.
表4 反应温度和反应时间对脱保护反应的影响
Table 4
| 编号 | 反应温度/℃ | 反应时间/h | HPLC含量/% | ||
|---|---|---|---|---|---|
| 化合物4 | 化合物3 | 杂质C | |||
| 1 | 20 | 1 | 33.25 | 65.92 | 0.05 |
| 2 | 20 | 5 | 97.62 | 0.74 | 0.47 |
| 3 | 20 | 22 | 97.58 | 0.29 | 1.20 |
| 4 | 30 | 1 | 97.63 | 0.29 | 1.06 |
| 5 | 30 | 5 | 97.32 | 0.43 | 1.37 |
| 6 | 40 | 1 | 97.32 | 0.35 | 1.42 |
| 7 | 40 | 5 | 90.31 | 2.21 | 6.74 |
| 8 | 50 | 1 | 94.19 | 1.20 | 3.74 |
| 9 | 60 | 1 | 85.06 | 3.00 | 11.07 |
3 结论
本文在达罗他胺合成的第一步Suzuki偶联和第二步水解脱保护反应中,分离纯化得到3个杂质A、B和C,借助HRMS和一维NMR确定了杂质A和B的结构,其中A为化合物2的脱硼酸频哪醇酯产物,而B为化合物1的双偶联产物. 借助HRMS、一维和二维NMR,特别是1H-13C HMBC谱,确定了化合物3和杂质C的结构,并完整归属了这两个同分异构体的谱图信号,同时对杂质C的形成过程及规避措施进行了分析和研究. 实验结果为达罗他胺的药物合成工艺研究提供了有益的科学参考.
利益冲突
无
附件材料(可在《波谱学杂志》期刊官网 http://magres.wipm.ac.cn 获取)
图S1 化合物3合成过程中反应液的HPLC谱图及各组分分布
图S2 化合物4合成过程中反应液的HPLC谱图及各组分分布
图S3 杂质A的1H NMR和13C NMR谱图
图S4 杂质B的1H NMR和13C NMR谱图
图S5 化合物4的1H NMR和13C NMR谱图
图S6 化合物4粗品的HPLC谱图及各组分分布
图S7 化合物4粗品的HPLC谱图及各组分分布(提高反应温度,延长反应时间)
图S8 化合物3纯化后的HPLC谱图及各组分分布
图S9 (a)化合物3和(b)杂质C的HRMS谱图
图S10 (a)化合物3和(b)杂质C的1H NMR谱图
图S11 化合物3的1H-1H NOESY谱图
图S12 杂质C的1H-1H NOESY谱图
图S13 (a)化合物3和(b)杂质C的13C NMR谱图
参考文献
Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods
[J].
DOI:10.1002/ijc.31937
PMID:30350310
[本文引用: 1]

Estimates of the worldwide incidence and mortality from 36 cancers and for all cancers combined for the year 2018 are now available in the GLOBOCAN 2018 database, compiled and disseminated by the International Agency for Research on Cancer (IARC). This paper reviews the sources and methods used in compiling the cancer statistics in 185 countries. The validity of the national estimates depends upon the representativeness of the source information, and to take into account possible sources of bias, uncertainty intervals are now provided for the estimated sex- and site-specific all-ages number of new cancer cases and cancer deaths. We briefly describe the key results globally and by world region. There were an estimated 18.1 million (95% UI: 17.5-18.7 million) new cases of cancer (17 million excluding non-melanoma skin cancer) and 9.6 million (95% UI: 9.3-9.8 million) deaths from cancer (9.5 million excluding non-melanoma skin cancer) worldwide in 2018.© 2018 UICC.
MicroRNA-194 promotes prostate cancer metastasis by inhibiting SOCS2
[J].
DOI:10.1158/0008-5472.CAN-16-2529
PMID:28011622
[本文引用: 1]
Serum levels of miR-194 have been reported to predict prostate cancer recurrence after surgery, but its functional contributions to this disease have not been studied. Herein, it is demonstrated that miR-194 is a driver of prostate cancer metastasis. Prostate tissue levels of miR-194 were associated with disease aggressiveness and poor outcome. Ectopic delivery of miR-194 stimulated migration, invasion, and epithelial-mesenchymal transition in human prostate cancer cell lines, and stable overexpression of miR-194 enhanced metastasis of intravenous and intraprostatic tumor xenografts. Conversely, inhibition of miR-194 activity suppressed the invasive capacity of prostate cancer cell lines and Mechanistic investigations identified the ubiquitin ligase suppressor of cytokine signaling 2 (SOCS2) as a direct, biologically relevant target of miR-194 in prostate cancer. Low levels of correlated strongly with disease recurrence and metastasis in clinical specimens. SOCS2 downregulation recapitulated miR-194-driven metastatic phenotypes, whereas overexpression of a nontargetable SOCS2 reduced miR-194-stimulated invasion. Targeting of SOCS2 by miR-194 resulted in derepression of the oncogenic kinases FLT3 and JAK2, leading to enhanced ERK and STAT3 signaling. Pharmacologic inhibition of ERK and JAK/STAT pathways reversed miR-194-driven phenotypes. The GATA2 transcription factor was identified as an upstream regulator of miR-194, consistent with a strong concordance between GATA2 and miR-194 levels in clinical specimens. Overall, these results offer new insights into the molecular mechanisms of metastatic progression in prostate cancer..©2016 American Association for Cancer Research.
Clinical development of darolutamide: a novel androgen receptor antagonist for the treatment of prostate cancer
[J].DOI:10.1016/j.clgc.2018.07.017 URL [本文引用: 1]
Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies
[J].
DOI:10.1038/srep12007
[本文引用: 1]
Activation of androgen receptor (AR) is crucial for prostate cancer growth. Remarkably, also castration-resistant prostate cancer (CRPC) is dependent on functional AR and several mechanisms have been proposed to explain the addiction. Known causes of CRPC include gene amplification and overexpression as well as point mutations of AR. We report here the pharmacological profile of ODM-201, a novel AR inhibitor that showed significant antitumor activity and a favorable safety profile in phase 1/2 studies in men with CRPC. ODM-201 is a full and high-affinity AR antagonist that, similar to second-generation antiandrogens enzalutamide and ARN-509, inhibits testosterone-induced nuclear translocation of AR. Importantly, ODM-201 also blocks the activity of the tested mutant ARs arising in response to antiandrogen therapies, including the F876L mutation that confers resistance to enzalutamide and ARN-509. In addition, ODM-201 reduces the growth of AR-overexpressing VCaP prostate cancer cells both in vitro and in a castration-resistant VCaP xenograft model. In contrast to other antiandrogens, ODM-201 shows negligible brain penetrance and does not increase serum testosterone levels in mice. In conclusion, ODM-201 is a potent AR inhibitor that overcomes resistance to AR-targeted therapies by antagonizing both overexpressed and mutated ARs. ODM-201 is currently in a phase 3 trial in CRPC.
New drug for the treatment of prostate cancer-darolutamide
[J].
DOI:10.3870/j.issn.1004-0781.2020.03.033
[本文引用: 1]
前列腺癌(PC)是发生在前列腺上皮组织的恶性肿瘤,也是男性泌尿生殖系统最常见、病死率最高的恶性肿瘤。前列腺肿瘤进展缓慢,早期不易发觉,未转移前,可有效诊疗和控制,一旦快速生长或扩散,病情严重恶化,转移后就不可治愈。据统计,2018年全球有近130万例新发病例,死亡35.9万例,占男性恶性肿瘤发病率的13.5%,居第3位;病死率占男性恶性肿瘤的6.7%,居第7位。发达国家PC发病率占总发病率的70%,随着发展中国家经济高速增长,近年来PC发病率也快速增长。中国2012年和2017年PC患病人数分别为82.9万例和121.2万例, 预计2022年将达到157.4万例,占全世界总发病率的6.62%,居第7位。达洛鲁胺(darolutamide)由德国拜耳(Bayer)制药公司研制,2014年6月拜耳公司与芬兰Orion制药公司签署联合开发协议。拜耳公司享有全球范围内推动该药商业化的独占权,Orion公司负责在欧洲共同推广该产品,并为全球市场生产提供药品。2019年3月11日,拜耳公司同时向美国食品药品管理局(FDA)、欧洲药品管理局(EMA)和日本厚生劳动省(MHLW)递交达洛鲁胺薄膜包衣片新药上市申请(NDA)。FDA于2019年4月29日给予优先审评认定,并于2019年7月29日批准上市,薄膜包衣片商品名为Nubeqa<sup>®</sup>。达洛鲁胺是一种口服非甾体雄激素受体(AR)抑制药,适用于治疗非转移性去势抵抗性前列腺癌(nmCRPC)患者。Ⅲ期临床试验表明,达洛鲁胺联合雄激素剥夺治疗(androgen deprivation therapy,ADT),比安慰药联合ADT,显著延长中位无转移生存期(MFS),分别为40.4个月与18.4个月,差异有统计学意义,并能将疾病转移或死亡的风险显著降低59%,且有良好的安全性。该文对达洛鲁胺薄膜包衣片的非临床和临床药理毒理学、临床研究、不良反应、适应证、剂量与用法、用药注意事项及知识产权状态和国内外研究进展等进行介绍。
治疗前列腺癌新药—达洛鲁胺(darolutamide)
[J].
DOI:10.3870/j.issn.1004-0781.2020.03.033
[本文引用: 1]
前列腺癌(PC)是发生在前列腺上皮组织的恶性肿瘤,也是男性泌尿生殖系统最常见、病死率最高的恶性肿瘤。前列腺肿瘤进展缓慢,早期不易发觉,未转移前,可有效诊疗和控制,一旦快速生长或扩散,病情严重恶化,转移后就不可治愈。据统计,2018年全球有近130万例新发病例,死亡35.9万例,占男性恶性肿瘤发病率的13.5%,居第3位;病死率占男性恶性肿瘤的6.7%,居第7位。发达国家PC发病率占总发病率的70%,随着发展中国家经济高速增长,近年来PC发病率也快速增长。中国2012年和2017年PC患病人数分别为82.9万例和121.2万例, 预计2022年将达到157.4万例,占全世界总发病率的6.62%,居第7位。达洛鲁胺(darolutamide)由德国拜耳(Bayer)制药公司研制,2014年6月拜耳公司与芬兰Orion制药公司签署联合开发协议。拜耳公司享有全球范围内推动该药商业化的独占权,Orion公司负责在欧洲共同推广该产品,并为全球市场生产提供药品。2019年3月11日,拜耳公司同时向美国食品药品管理局(FDA)、欧洲药品管理局(EMA)和日本厚生劳动省(MHLW)递交达洛鲁胺薄膜包衣片新药上市申请(NDA)。FDA于2019年4月29日给予优先审评认定,并于2019年7月29日批准上市,薄膜包衣片商品名为Nubeqa<sup>®</sup>。达洛鲁胺是一种口服非甾体雄激素受体(AR)抑制药,适用于治疗非转移性去势抵抗性前列腺癌(nmCRPC)患者。Ⅲ期临床试验表明,达洛鲁胺联合雄激素剥夺治疗(androgen deprivation therapy,ADT),比安慰药联合ADT,显著延长中位无转移生存期(MFS),分别为40.4个月与18.4个月,差异有统计学意义,并能将疾病转移或死亡的风险显著降低59%,且有良好的安全性。该文对达洛鲁胺薄膜包衣片的非临床和临床药理毒理学、临床研究、不良反应、适应证、剂量与用法、用药注意事项及知识产权状态和国内外研究进展等进行介绍。
Graphical synthetic routes to darolutamide
[J].
达罗他胺合成路线图解
[J].
Androgen receptor modulating compounds: WO2011051540A1
[P]. 2010-10-27.
Androgen receptor modulating carboxamides: WO2012143599A1
[P]. 2012-10-26.
Discovery and biological evaluation of darolutamide derivatives as inhibitors and down-regulators of wild-type AR and the mutants
[J].DOI:10.1016/j.ejmech.2019.111608 URL [本文引用: 3]
Structure characterization and analgesic activity of novel pyrazolo[3,4-d]pyrimidin-4-one Derivatives
[J].
新型吡唑并[3,4-d]嘧啶-4-酮类衍生物的结构表征和镇痛活性
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}