Comparison of Different Approaches for Estimation of the Detection Limit of Quantitative NMR
CHEN Lei,, LIU Hong-bing, LIU Hui-li
State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, National Center for Magnetic Resonance in Wuhan, Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences, Wuhan 430071, China
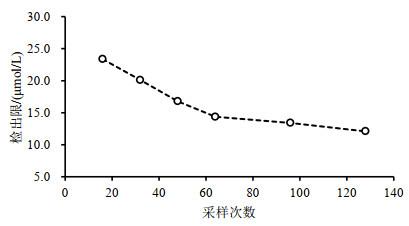
在对核磁共振(Nuclear Magnetic Resonance,NMR)定量检测进行方法验证时,检出限是一项重要的指标.本文对文献中报道的校准曲线法、校准方程参数法、数学模型法、单浓度点评估法进行了系统梳理,重点介绍了这些方法的基本原理、计算公式和特点,并针对信噪比法的不足,提出了信噪比回归曲线法.采用上述五种方法计算了水溶液中甲酸钠1H NMR检测时的检出限,并讨论了采样次数对检出限的影响.在700 MHz核磁共振谱仪上,当采样次数为64时,甲酸钠的检出限在10.4~14.4 μmol/L之间.该结果为1H NMR检出限的评估提供了参考.
关键词:液体核磁共振
;
定量检测
;
检出限
;
信噪比回归曲线法
Abstract
Limit of detection (LOD), which indicates the detection ability of an analytical method, is an important parameter used in validating quantitative 1H nuclear magnetic resonance (NMR) method. Different approaches to evaluate LOD have been reported in literature, including the calibration curve approach, regression parameters-based approach, ASTM approach (proposed by American Society of Testing Materials), EPA approach (established by the United States Environmental Protection Agency) and the signal-to-noise ratio approach. In this study, systemic analyses and summaries of all mentioned approaches were given together with their principles, equations and characteristics. A novel approach based on signal-to-ratio regression curve for determining the detection limit was proposed, which can overcome weaknesses of the signal-to-noise ratio approach. The LOD of liquid-state 1H NMR method in the determination of sodium formate in aqueous solution was calculated using 5 different approaches. The influence of the number of scans on LOD was discussed. The results showed that the LOD was in the range of 10.4~14.4 μmol/L when the number of scans was 64 with 700 MHz NMR spectrometer. In conclusion, the study can provide a reference for determining the LOD of 1H quantitative NMR.
Keywords:liquid-state nuclear magnetic resonance
;
quantitative detection
;
limit of detection (LOD)
;
signal-to-noise regression approach
CHEN Lei. Comparison of Different Approaches for Estimation of the Detection Limit of Quantitative NMR. Chinese Journal of Magnetic Resonance[J], 2022, 39(2): 230-242 doi:10.11938/cjmr20212944
引言
检出限(Limit of Detection,LOD)是评价一种分析方法检测性能的重要指标,特别是反映了对低浓度样品的检测能力.分析方法验证、标准制定、实验室检测能力确认都会涉及到检出限的评估.因此,研究人员对检出限进行了长期的研究和探讨,提出了多种检出限评估方法,并形成了一些标准.
1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14].
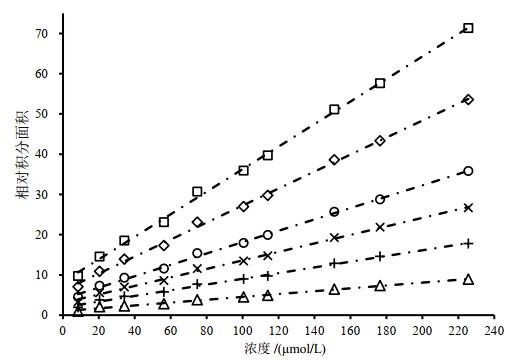
Fig.2
Scatter plots of sodium formate concentration vs. relative integral area and calibration curves with various numbers of scan (16 (△), 32 (+), 48 (×), 64 (○), 96 (◇) and 128 (□))
信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性.
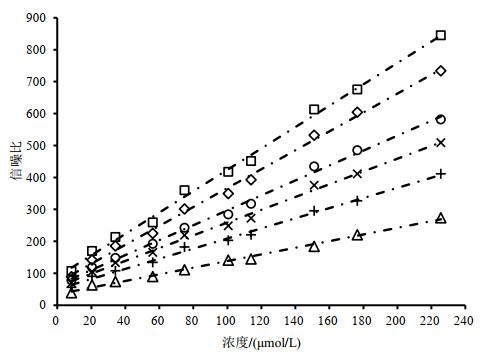
Fig.5
Scatter plots of sodium formate concentration vs. signal-to-noise ratio and regression curves with various numbers of scan (16 (△), 32 (+), 48 (×), 64 (○), 96 (◇) and 128 (□))
Table 3
表3
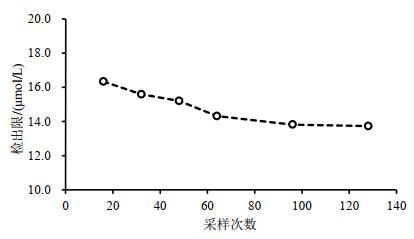
表3不同采样次数下,信噪比回归曲线法中的拟合参数及计算出的检出限
Table 3 Fitting parameters and limits of detection calculated by the signal-to-noise ratio regression method with different numbers of scan
Technical Committee ISO/TC 69, Applications of statistical methods, Subcommittee SC 6, Measurement methods and results. Capability of detection - Part 2: Methodology in the linear calibrations case: ISO 11843-2: 2000[S]. Geneva: International Organization for Standardization, 2000.
ASTM Committee D19 on Water, Subcommittee D19.02 on quality systems, specification, and statistics. standard practice for 99%/95% inter-laboratory detection estimate (IDE) for analytical methods with negligible calibrations error: ASTM D6091-07(Reapproved 2014)[S]. West Conshohocken: ASTM International, 2014.
US-EPA. 40 CFR Appendix B to part 136-definition and procedure for the determination of the method detection limit, revision 2: EPA 821-R-16-006[S]. Washington DC: US Environmental Protection Agency, 2016
A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques (Technical Report)
INTERNATIONAL CONFERENCE ON HARMONIZATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE. ICH harmonized tripartite guideline: Validation of analytical procedures: Text and Methodology Q2(R1)[S]. Geneva: ICH, 2005.
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
7
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性. ...
3
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 定量质子核磁共振(1H Nuclear Magnetic Resonance,1H NMR)具有样品制备简单、操作方便快捷、重复性好的特点,广泛应用于化工、天然产物、食品、医药等研究领域[26-28],已被《英国药典》[29]、《中国药典》[8]收录.在建立定量1H NMR检测方法时,和准确度、精密度一样,检出限也是一项重要的衡量指标.但一些定量NMR文献中仅给了检出限的数值,既没有指明评估采用的方法,也没有给出具体过程,不够严谨.同时部分文章对检出限的理解不够,导致在评估检出限时有偏差[30].文献中关于NMR检出限的对比分析也比较少,例如评估方法、计算过程、计算结果的对比,以及使用条件、注意事项等. ...
... 信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性. ...
Trace analyses for wastewaters
2
1981
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques (Technical Report)
1
1997
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
检出限几种常见计算方法的分析和比较
0
2010
检出限几种常见计算方法的分析和比较
0
2010
A novel approach for the determination of detection limits for metal analysis of environmental water samples
1
2005
... 1998年,国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)在《分析术语纲要》[1]中规定检出限以浓度(或质量)表示,是指根据特定的分析步骤能够合理检测出的最小分析信号求得的最低浓度(或质量),并给出了计算公式.对空白样品响应信号的多次测定并得到响应值的标准偏差,是其关键步骤.国际标准化组织(International Standardization Organization,ISO)制定了基于校准曲线法的评估标准(ISO 11843.2—2000)[2],国家标准GB/T 17378.2—2007[3]和GB/T 33260.2—2018[4]中也采纳了该方法.国家标准GB/T 27417—2017[5]中指出检出限可以分为仪器检出限和方法检出限:仪器检出限是指用仪器可靠地将目标分析物信号从背景(噪音)中识别出来时,分析物的最低浓度或量;方法检出限定义为用特定方法可靠地将分析物待测定信号从特定基质背景中识别或区分出来时,分析物的最低浓度或量.方法检出限是前处理过程和仪器检测能力的综合反映,考虑了基质的影响.仪器检出限和方法检出限采用相同的方法进行计算.在实际检测中,如果检测结果低于检出限,应按“未检出”出具结论,并报告检出限的量值.该国标中列出了一种利用校准曲线回归方程的参数评估检出限的方法.该方法可以认为是校准曲线法的简化,虽然理论基础薄弱了一些,但优点是计算相对简便.美国材料实验协会(American Society of Testing Materials,ASTM)根据数学模型法提出了实验室间检出限的评估方法(ASTM D6091—07)[6],国家标准GB/T 27415—2013[7]等效采用了ASTM标准.在药品分析领域,《中国药典》(2020版)“分析方法验证指导原则”中规定了检出限的3种评估方法[8]:直观法、信噪比法、基于响应值标准偏差和标准曲线斜率法.美国环境保护署(United States Environmental Protection Agency,USEPA)基于单浓度水平,建立了一套检出限评价方法,称之为单浓度水平评估法(EPA法)[9, 10].国家生态环境标准HJ 168—2020[11]等效采用了此方法.在上述评估方法的基础上,研究人员针对不同的体系、采样不同的理论,又提出了一些新的方案,例如上限法、极差法等[12-14]. ...
... 信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性. ...
Limits for qualitative detection and quantitative determination: Application to radiochemistry
1
1968
... 信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性. ...
Over a century of detection and quantification capabilities in analytical chemistry - historical overview and trends
1
2014
... 信噪比法被人用药品注册技术要求国际协调委员会(International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)发布的“分析方法指导原则”收录[33].《中国药典》(2020版)[8]和国家标准GB/T 27417—2017[5]也采纳其作为检出限的评估方法.信噪比法适用于能显示基线噪声的仪器分析方法,通过已知低浓度目标化合物的响应信号与基线噪声比较,确定出能被可靠检出的目标化合物的最低浓度或者量.一般以信噪比为3:1或2:1时相应的浓度或者量作为检出限,在色谱、质谱分析方法中,评估检出限较常采用.检出限是一个统计学的概念[34, 35],前面提及的方法都是从统计理论的不同方面出发,推导出检出限的公式.而信噪比法是通过实验测量信噪比,再确定检出限.这使得“检出限”成为了一个实验值,不太符合检出限本身统计学定义的特性.同时使用该方法时需要配制一系列很低浓度的标准溶液,寻找信噪比为3或者2的响应信号.如果信噪比高于阈值,则继续配制更低浓度的标准溶液,需要反复多次尝试.当信噪比较低时,对应的响应信号强度弱,测量困难,容易受噪声波动的影响,导致相对标准差偏大,会削弱检出限评估的可靠性. ...






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}