1. School of Engineering, Macquarie University, Sydney, New South Wales 2109
2. Laboratory for Catalysis Engineering, School of Chemical and Biomolecular Engineering & Sydney Nano Institute, The University of Sydney, Sydney, NSW 2006
Solid-State NMR Spectroscopy Studies of Enhanced Acidity of Silica-Aluminas Based on Penta-Coordinated Aluminum Species
WANG Zi-chun1, HUANG Jun2, JIANG Yi-jiao,1
1. School of Engineering, Macquarie University, Sydney, New South Wales 2109, Australia
2. Laboratory for Catalysis Engineering, School of Chemical and Biomolecular Engineering & Sydney Nano Institute, The University of Sydney, Sydney, NSW 2006, Australia
Australian Research Council Discovery Projects. DP150103842 Australian Research Council Discovery Projects. DP180104010 Australian Research Council Discovery Projects. DE190101618
Brønsted acid sites (BASs) on amorphous silica-aluminas (ASAs) are key active sites that can be generated based on tetra-coordinated aluminum (AlIV) species, albeit much weaker than that of zeolites. Penta-coordinated aluminum (AlV) species are recently reported to enhance the acidity of ASAs, and to overcome the limitations of AlIV species. This review introduces novel strategies to control the synthesis of AlV-enriched ASAs using flame spray pyrolysis (FSP) techniques. The acidic properties and local structure of AlV-enriched ASAs were characterized by various advanced 2D solid-state nuclear magnetic resonance (SSNMR) techniques and in situ1H MAS NMR experiments, showing two new types of BASs from moderate to zeolitic acid strength based on AlV species. 27Al and 1H MAS NMR studies uncovered the role of AlV species in tailoring single-atom catalysts on ASAs.
WANG Zi-chun. Solid-State NMR Spectroscopy Studies of Enhanced Acidity of Silica-Aluminas Based on Penta-Coordinated Aluminum Species. Chinese Journal of Magnetic Resonance[J], 2021, 38(4): 552-570 doi:10.11938/cjmr20212930
Fig.1
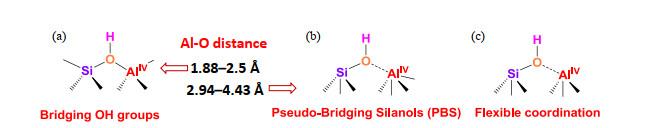
Proposed models for Brønsted acid sites (BASs) in silica-alumina catalysts[7]. (a) BAS consisting of a bridging silanol site bonded to AlIV site (SiOHAl) in zeolites; (b) BAS consisting of PBS interacting with AlIV site; (c) BAS consisting of the flexible coordination between silanol oxygen and neighbouring AlIV. In the latter two cases, the dotted line does not denote a covalent bond but only the close proximity between O and Al atoms
图2
(a, b) SA/10和(c, d) SA/30的(a, c) 723 K下脱水后及(b, d)再吸水后的27Al多量子魔角旋转(MQMAS)谱(16.4 T);以及(e, f) SA/10和(g, h) SA/30相应的能量色散X射线光谱(EDX)[6].(i) SA/10的三维原子探针层析成像分析(3D-APT)[8]
Fig.2
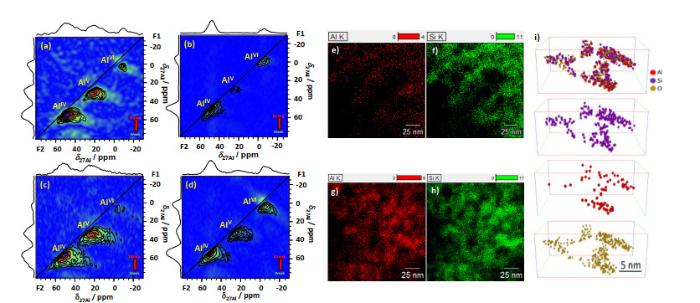
27Al MQMAS spectra of SA/10 (a, b) and SA/30 (c, d) after dehydrated at 723 K (a, c) and after rehydration (b, d), recorded at a magnetic field of 16.4 T, and corresponding EDX images of SA/10 (e, f) and SA/30 (g, h)[6]. (i) 3D-APT reconstruction of isolated SA/10 nanoparticles[8]
理论计算认为AlV能够与邻近的SiOH产生相互作用,从而促进表面B酸位的形成[15].但由于缺乏AlV与SiOH相互作用的相关证据,该假设存在广泛争议.首先,常用的一维27Al MAS NMR谱,如1H/27Al双共振实验(TRAPDOR)与27Al{1H}交叉极化(CP)实验,能够提供质子与邻近Al原子的作用信息,但是不能用于区分Al原子的配位[37].二维NMR相关谱广泛用于研究沸石中B酸位的微观结构,同时能够区分不同配位Al原子与邻近质子的相关性[38, 39].但是应用二维NMR相关谱研究ASAs的结构非常困难.这主要是由于ASAs中缺乏长程有序结构、铝的化学环境分布广泛,因而导致探测信号的灵敏度低且相互重叠,特别是难以实现对ASAs中含量极低的AlV的观测.
Fig.3
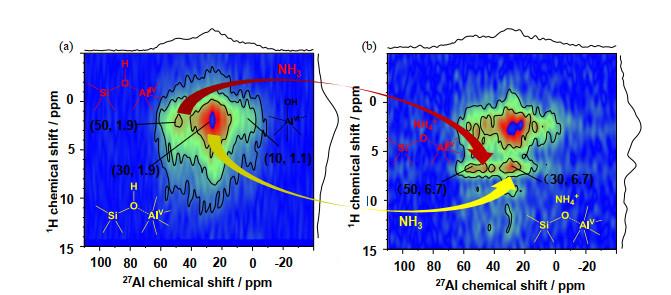
27Al-{1H} D-HMQC 2D spectra of SA/50. (a) SA/50 dehydrated at 723 K for 12 h under vacuum, and (b) after ammonia loading and evacuated at 373 K for 1 h. The spectra were recorded at 18.8 T with a MAS frequency of νR=20 kHz, and τrec=1.0 ms for dehydrated and τrec=900 μs for ammonia-loaded sample, respectively[7]
Fig.4
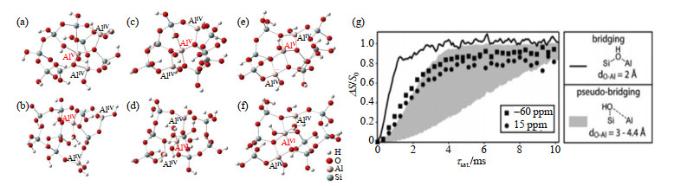
Optimized structure of AlV species in dehydrated states (a, c and e) and in corresponding rehydrated states (b, d, and f), both calculated at B3LYP/6-31g theoretical level[6]; (g) 17O{27Al} TRAPDOR curves obtained from 17O{1H, 27Al} PRESTO-TRAPDOR experiment were simulated to determine the Al-OH distance, for discriminating the structure of AlV-BASs, corresponding to bridging silanols (black line, O-Al distance of 2 Å) and pseudo-bridging silanols (gray shaded area, O-Al distance range of 3 to 4.4 Å)[41], the corresponding 17O chemical shifts were at -60 and 15 ppm
Fig.6
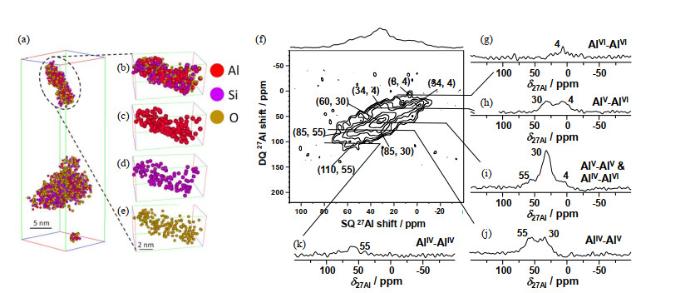
(a)~(e) 3D APT reconstruction of two isolated SA/50 nanoparticles showing all atoms; (f) 27Al DQ-SQ 2D NMR spectrum recorded at 18.8 T with νR=20 kHz of dehydrated SA/50, and (g)~(k) rows extracted from the 2D spectrum corresponding to the various autocorrelation and cross-peaks[8]
Fig.7
Proposed models for BAS on ASAs generated by (a) one Al center per SiOH for moderate BAS, and (b) two Al centers per SiOH group, leading to zeolitic acid strengths[8]
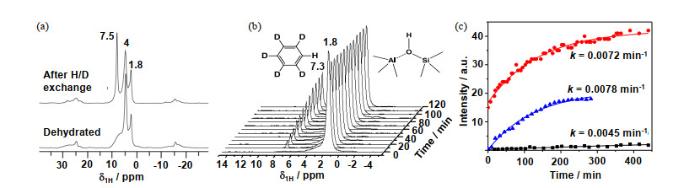
固体酸催化剂中的B酸位能够将质子传递给吸附的碳氢化合物分子,形成碳正离子.碳正离子是碳氢化合物转化的重要反应中间体.通常,强B酸位有助于碳正离子的形成.如沸石中的强酸性桥式羟基能够有效活化苯环上的C-H键,显著提升芳烃的烷基化反应效率[48-51].通过原位1H MAS NMR谱能够研究脱水固体酸表面各种羟基质子与氘代芳烃的相互作用(氢氘交换).通过拟合参与氢氘交换反应的芳烃浓度与时间的关系,可以得到反应的速率曲线.结合不同温度下的速率曲线能够给出活化能等衡量固体酸催化活性的信息.
Wang等[8]通过原位1H MAS NMR谱对比氘代苯与H-ZSM-5沸石及AlV富集的ASAs表面B酸位之间的氢氘反应速率,证明了ASAs中存在强B酸位.与文献报道[48-51]相似,H-ZSM-5沸石表面的酸性羟基(δH 4)含量大幅降低而中性SiOH(δH 1.8)含量基本不变,同时苯环上的质子(δH 7.5)含量显著提高[图 8(a)].这说明了沸石中的酸性桥式羟基是该反应的主要活性位,而中性SiOH不参与反应.但是氢氘反应前后,SA/50表面的SiOH(δH 1.8)含量大幅降低并伴随着苯环上的质子(δH 7.3~7.5)含量显著提高[图 8(b)],这一现象说明SA/50表面的酸性SiOH同样能够催化该反应.通过拟合能够得到了氘代苯与不同固体酸表面B酸位反应的速率k [图 8(c)].一般来说,速率k与B酸位的强度呈正比.该实验中,研究人员通过氘代苯与表面B酸位的1:1吸附及低温扩散后进行测试,更进一步降低了苯的浓度及扩散对实验的影响.该研究表明SA/50与H-ZSM-5沸石具有几乎相同的速率k,远高于SA/10.这说明了SA/50与H-ZSM-5沸石具有相似的B酸强度,DQ-SQ 2D NMR谱以及SA/50的1H MAS NMR谱中没有观测到酸性桥式羟基,SA/50中的强B酸与采用2-13C-丙酮位探针分子的13C CP/MAS NMR谱表征结果[10]一致.结合27Al位被认为是多个不饱和Al原子与同一个SiOH协同作用的结果.这种强B酸位的形成能够显著增强ASAs在一系列C-H键活化反应中的催化活性.
图8
原位1H MAS NMR谱(9.4 T)研究313 K时氘代苯与固体酸表面羟基的氢氘交换.(a) H-ZSM-5沸石和(b) SA/50的1H MAS NMR谱图;(c)采用H-ZSM-5(上)、SA/50(中)和SA/10(下)分别拟合得到的氢氘交换速率曲线[8]
Fig.8
Catalytic performance of H-ZSM-5 and AlV-enriched ASAs in H/D exchange with d6-benzene at 313 K. 1H MAS NMR spectra at 9.4 T of (a) zeolite H-ZSM-5 and (b) SA/50 (stack plot spectra). (c) Kinetics and H/D exchange rates k for H-ZSM-5 (top), SA/50 (middle), and SA/10 (bottom)[8]
Fig.9
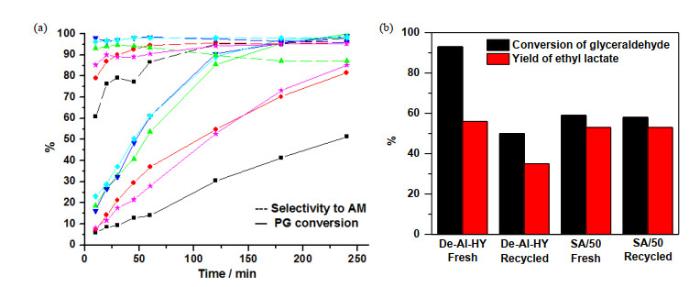
Catalytic conversion of C3 sugars over ASAs. (a) Catalytic conversion of phenylglyoxal (―) and selectivity to alkyl mandelate (---) as a function of reaction time in MeOH (■), EtOH (●), i-PrOH (▼), n-PrOH (◆), n-BuOH (▲) over SA/70 and reaction in i-PrOH (★) over De-Al-HY[26]. (b) Catalytic glyceraldehyde conversion in ethanol over fresh De-Al-HY and SA/50 and after five recycle uses. Conditions: 1.25 mL of alcohol solution containing 0.4 mol/L phenylglyoxal or glyceraldehyde, 0.05 g catalyst, at 363 K for 6 h with stirring[8]
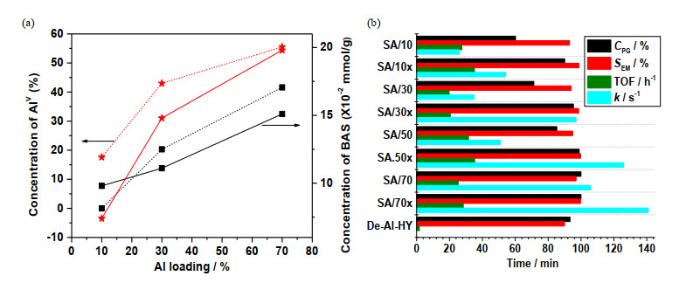
在相同条件下,使用二甲苯为溶剂能够产生更高的火焰温度和更快的冷却速度.通过对得到的SA/Xx的27Al MAS NMR谱进行模拟及定量分析,发现以二甲苯为溶剂合成的ASAs能够显著提高AlV的含量[如图 10(a)].与使用甲醇/乙酸(体积比1:1)混合溶剂合成的SA/X相比,使用二甲苯提高AlV含量能够促进AlV与SiOH相互作用,将B酸含量提高了近30%.与传统AlIV富集的ASAs相比,能够将B酸含量提高近10倍[61].特别是使用具有更高燃烧熵的溶剂不仅能够提高AlV含量,还能改善AlV在氧化硅中的分布,促进B酸的形成,因而能在大幅降低铝的用量的条件下合成具有相似酸性的ASAs(如SA/30x和SA/70)[9].
Fig.10
(a) Correlation between the concentration of Brønsted acidic OH groups (BAS, solid line) and AlV species (dashed line) as a function of the Al content in dehydrated FSP ASAs prepared by using methanol/acetic acid (1:1 by volume, ■) and xylene (★), respectively; (b) Catalytic conversion of PG in ethanol. The PG conversion (CPG), EM selectivity (SEM), reaction rates k, and TOFs were obtained from Ref. [9], respectively. Same reaction conditions as shown in Fig. 9
Fig.11
27Al MAS NMR spectra of (a) ASA, (b) Pt/ASA, (c) Al2O3 (yellow dash line), and Pt/Al2O3 (blue solid line). Static 27Al NMR of wet samples obtained after: (d) H2PtCl6 mixed with ASA, (e) mixing (d) with NH3•H2O, and (f) after heating mixture (e) at 90 ℃ for 1 h, recorded at 11.7 T[27]
Fig.12
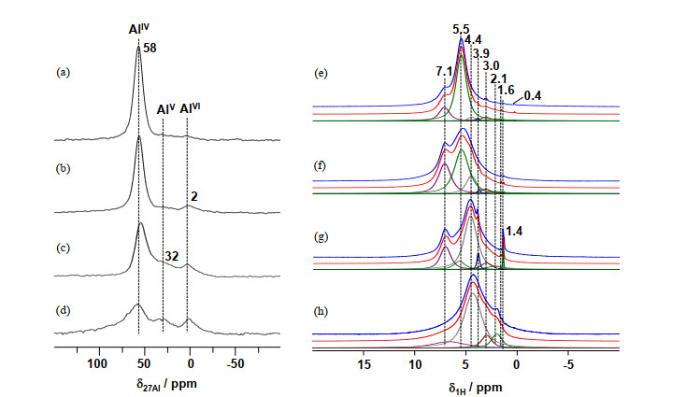
27Al (a~d) and 1H (e~h) MAS NMR spectra of the as-synthesized Pt/ASA dried at different temperatures: room temperature (Pt/ASA-RT) (a, e), 100 ℃ (b, f), 200 ℃ (c, g) and 350 ℃ (d, h) [27]
Solid-state nuclear magnetic resonance spectroscopy-based analysis of local structure, acidic property, and activity for solid acids solid-state nuclear magnetic resonance spectroscopy-based analysis of local structure and acidic property for solid acids
Mechanically induced phase transformation of gamma-Al2O3 into alpha-Al2O3. Access to structurally disordered gamma-Al2O3 with a controllable amount of pentacoordinated Al sites
Atomic description of the interface between silica and alumina in aluminosilicates through dynamic nuclear polarization surface-enhanced NMR spectroscopy and first-principles calculations
Extra-framework aluminium species in hydrated faujasite zeolite as investigated by two-dimensional solid-state NMR spectroscopy and theoretical calculations
Magic-angle spinning nuclear-magnetic-resonance studies of water-molecules adsorbed on Brønsted-acid and Lewis-acid sites in zeolites and amorphous silica-aluminas
... [6].(i) SA/10的三维原子探针层析成像分析(3D-APT)[8]<sup>27</sup>Al MQMAS spectra of SA/10 (a, b) and SA/30 (c, d) after dehydrated at 723 K (a, c) and after rehydration (b, d), recorded at a magnetic field of 16.4 T, and corresponding EDX images of SA/10 (e, f) and SA/30 (g, h)<sup>[<xref ref-type="bibr" rid="b6">6</xref>]</sup>. (i) 3D-APT reconstruction of isolated SA/10 nanoparticles<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.2
AlV富集的ASAs中不同铝配位及B酸的浓度[6, 9, 10] ...
... [6]. (i) 3D-APT reconstruction of isolated SA/10 nanoparticles[8]Fig.2
AlV富集的ASAs中不同铝配位及B酸的浓度[6, 9, 10] ...
... AlV富集的ASAs中不同铝配位及B酸的浓度[6, 9, 10] ...
... Al concentration and population density of BASs in AlV-enriched ASAs[6, 9, 10] ...
... [6];(g)通过17O{1H, 27Al} PRESTO-TRAPDOR实验及对得到的17O{27Al} TRAPDOR曲线进行模拟,确定ASAs中Al-OH的键长用以区分AlV-BAS的结构,如酸性桥式羟基(黑色曲线,O-Al键长为2 Å)或酸性伪桥式硅羟基(灰色区域,O-Al键长为3~4.4 Å)[41],“−60 ppm”和“15 ppm”为17O NMR信号 Optimized structure of Al<sup>V</sup> species in dehydrated states (a, c and e) and in corresponding rehydrated states (b, d, and f), both calculated at B3LYP/6-31g theoretical level<sup>[<xref ref-type="bibr" rid="b6">6</xref>]</sup>; (g) <sup>17</sup>O{<sup>27</sup>Al} TRAPDOR curves obtained from <sup>17</sup>O{<sup>1</sup>H, <sup>27</sup>Al} PRESTO-TRAPDOR experiment were simulated to determine the Al-OH distance, for discriminating the structure of Al<sup>V</sup>-BASs, corresponding to bridging silanols (black line, O-Al distance of 2 Å) and pseudo-bridging silanols (gray shaded area, O-Al distance range of 3 to 4.4 Å)<sup>[<xref ref-type="bibr" rid="b41">41</xref>]</sup>, the corresponding <sup>17</sup>O chemical shifts were at -60 and 15 ppmFig.43 Al<sup>V</sup>富集的ASAs中强B酸位形成的多铝中心协同作用模型
... [6]; (g) 17O{27Al} TRAPDOR curves obtained from 17O{1H, 27Al} PRESTO-TRAPDOR experiment were simulated to determine the Al-OH distance, for discriminating the structure of AlV-BASs, corresponding to bridging silanols (black line, O-Al distance of 2 Å) and pseudo-bridging silanols (gray shaded area, O-Al distance range of 3 to 4.4 Å)[41], the corresponding 17O chemical shifts were at -60 and 15 ppm Fig.43 Al<sup>V</sup>富集的ASAs中强B酸位形成的多铝中心协同作用模型
Br?nsted acid sites based on penta-coordinated aluminum species
6
2016
... 以硅铝固体酸,例如沸石、无定型硅铝(amorphous silica-aluminas,ASAs)材料为代表的固体酸催化剂广泛应用于石油炼制、煤化工、精细化工、药物合成、环境保护、新能源开发、新材料合成等领域[1-3],同时用以替代液体酸催化剂,实现绿色、连续的化工生产过程[4, 5].硅铝固体酸催化剂的催化活性及目标产物选择性主要取决于其表面酸性.Brønsted酸(简称B酸)是硅铝固体酸中主要的催化活性位,主要通过在中性或弱酸性的氧化硅中掺杂Al原子产生(见图 1).而沸石中的非骨架铝离子及ASAs催化剂中的氧化铝相表面缺陷位能够提供Lewis酸性(简称L酸).硅铝固体酸中B酸的结构<sup>[<xref ref-type="bibr" rid="b7">7</xref>]</sup>:(a)酸性桥式羟基;(b)酸性伪桥式硅羟基;(c) Al<sup>IV</sup>与SiOH通过静电作用形成的B酸位.后两个模型中,虚线仅代表原子间的接近关系而不代表成键关系Proposed models for Brønsted acid sites (BASs) in silica-alumina catalysts<sup>[<xref ref-type="bibr" rid="b7">7</xref>]</sup>. (a) BAS consisting of a bridging silanol site bonded to Al<sup>IV</sup> site (SiOHAl) in zeolites; (b) BAS consisting of PBS interacting with Al<sup>IV</sup> site; (c) BAS consisting of the flexible coordination between silanol oxygen and neighbouring Al<sup>IV</sup>. In the latter two cases, the dotted line does not denote a covalent bond but only the close proximity between O and Al atomsFig.1
... [7]. (a) BAS consisting of a bridging silanol site bonded to AlIV site (SiOHAl) in zeolites; (b) BAS consisting of PBS interacting with AlIV site; (c) BAS consisting of the flexible coordination between silanol oxygen and neighbouring AlIV. In the latter two cases, the dotted line does not denote a covalent bond but only the close proximity between O and Al atoms Fig.1
... [7].相应谱图在18.8 T下采集,相应参数为νR=20 kHz,脱水样品的τrec=1.0 ms,吸氨样品的τrec=900 μs <sup>27</sup>Al-{<sup>1</sup>H} <i>D</i>-HMQC 2D spectra of SA/50. (a) SA/50 dehydrated at 723 K for 12 h under vacuum, and (b) after ammonia loading and evacuated at 373 K for 1 h. The spectra were recorded at 18.8 T with a MAS frequency of <i>ν</i><sub>R</sub>=20 kHz, and <i>τ</i><sub>rec</sub>=1.0 ms for dehydrated and <i>τ</i><sub>rec</sub>=900 μs for ammonia-loaded sample, respectively<sup>[<xref ref-type="bibr" rid="b7">7</xref>]</sup>Fig.3
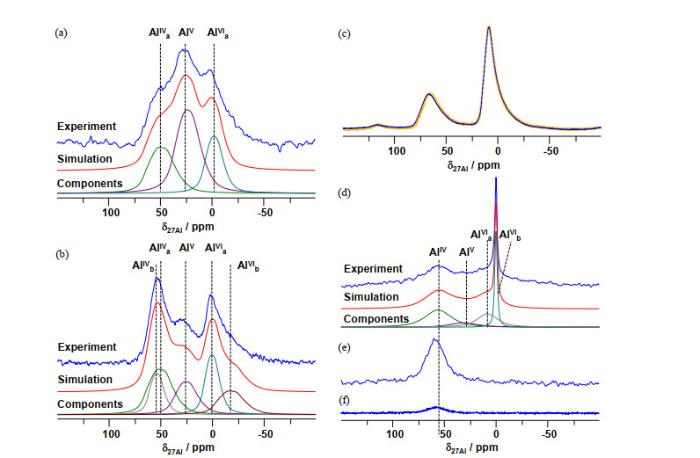
... 27Al多量子魔角旋转(MQMAS)谱常用来测定Al原子的配位及局部对称性[31].如图 2(a)和2(c)中,通过化学位移,在脱水SA/X的27Al MQMAS谱中确定了AlV、AlIV及微量AlVI的存在,并能够得到相应的NMR参数.用得到的NMR参数能够分解相应的一维27Al MAS NMR谱,从而得到不同配位Al的含量(表 1).在氧化铝及ASAs中,AlV的出现通常伴随着大量AlVI的存在[16, 32].因此,AlV通常被认为是氧化铝或者ASAs的氧化铝分相中少量具有氧缺陷的AlVI[33].但是,相关X射线衍射仪(X-ray diffraction,XRD)及高分辨透射电镜(high-resolution transmission electron microscopy,HRTEM)研究都说明SA/X中并不存在明显的铝分相[6].同时,在AlV富集的ASAs中仅观测到微量的AlVI(1 at.% ~ 7 at.%),而AlV的含量则高达(37 at.% ~ 55 at.%),这说明AlV的形成完全不依赖于ASAs中的铝分相[6].(a, b) SA/10和(c, d) SA/30的(a, c) 723 K下脱水后及(b, d)再吸水后的<sup>27</sup>Al多量子魔角旋转(MQMAS)谱(16.4 T);以及(e, f) SA/10和(g, h) SA/30相应的能量色散X射线光谱(EDX)<sup>[<xref ref-type="bibr" rid="b6">6</xref>]</sup>.(i) SA/10的三维原子探针层析成像分析(3D-APT)<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup><sup>27</sup>Al MQMAS spectra of SA/10 (a, b) and SA/30 (c, d) after dehydrated at 723 K (a, c) and after rehydration (b, d), recorded at a magnetic field of 16.4 T, and corresponding EDX images of SA/10 (e, f) and SA/30 (g, h)<sup>[<xref ref-type="bibr" rid="b6">6</xref>]</sup>. (i) 3D-APT reconstruction of isolated SA/10 nanoparticles<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.2
... [8] (a)~(e) 3D APT reconstruction of two isolated SA/50 nanoparticles showing all atoms; (f) <sup>27</sup>Al DQ-SQ 2D NMR spectrum recorded at 18.8 T with <i>ν</i><sub>R</sub>=20 kHz of dehydrated SA/50, and (g)~(k) rows extracted from the 2D spectrum corresponding to the various autocorrelation and cross-peaks<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.6
... [8] Proposed models for BAS on ASAs generated by (a) one Al center per SiOH for moderate BAS, and (b) two Al centers per SiOH group, leading to zeolitic acid strengths<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.7
... Wang等[8]通过原位1H MAS NMR谱对比氘代苯与H-ZSM-5沸石及AlV富集的ASAs表面B酸位之间的氢氘反应速率,证明了ASAs中存在强B酸位.与文献报道[48-51]相似,H-ZSM-5沸石表面的酸性羟基(δH 4)含量大幅降低而中性SiOH(δH 1.8)含量基本不变,同时苯环上的质子(δH 7.5)含量显著提高[图 8(a)].这说明了沸石中的酸性桥式羟基是该反应的主要活性位,而中性SiOH不参与反应.但是氢氘反应前后,SA/50表面的SiOH(δH 1.8)含量大幅降低并伴随着苯环上的质子(δH 7.3~7.5)含量显著提高[图 8(b)],这一现象说明SA/50表面的酸性SiOH同样能够催化该反应.通过拟合能够得到了氘代苯与不同固体酸表面B酸位反应的速率k [图 8(c)].一般来说,速率k与B酸位的强度呈正比.该实验中,研究人员通过氘代苯与表面B酸位的1:1吸附及低温扩散后进行测试,更进一步降低了苯的浓度及扩散对实验的影响.该研究表明SA/50与H-ZSM-5沸石具有几乎相同的速率k,远高于SA/10.这说明了SA/50与H-ZSM-5沸石具有相似的B酸强度,DQ-SQ 2D NMR谱以及SA/50的1H MAS NMR谱中没有观测到酸性桥式羟基,SA/50中的强B酸与采用2-13C-丙酮位探针分子的13C CP/MAS NMR谱表征结果[10]一致.结合27Al位被认为是多个不饱和Al原子与同一个SiOH协同作用的结果.这种强B酸位的形成能够显著增强ASAs在一系列C-H键活化反应中的催化活性. ...
... [8] Catalytic performance of H-ZSM-5 and Al<sup>V</sup>-enriched ASAs in H/D exchange with <i>d</i><sub>6</sub>-benzene at 313 K. <sup>1</sup>H MAS NMR spectra at 9.4 T of (a) zeolite H-ZSM-5 and (b) SA/50 (stack plot spectra). (c) Kinetics and H/D exchange rates <i>k</i> for H-ZSM-5 (top), SA/50 (middle), and SA/10 (bottom)<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.85 ASAs的酸性强化与高效生物质转化
... a: ASAs的比表面积(ABET)取自参考文献[10],De-Al-HY的ABET取自参考文献[48]. b: 扁桃酸乙酯的产率(YEM)、选择性(SEM)及转换频率(TOFs)均取自参考文献[26]. c: ASAs及De-Al-HY的酸位含量分别取自参考文献[10]和[48]ASAs上C3糖类的催化转化.(a) SA/70催化苯甲酰甲醛(―)与多种醇反应合成扁桃酸盐(---)的转化率/选择性—时间曲线:甲醇(■),乙醇(●),异丙醇(▼),正丙醇(◆),正丁醇(▲),及对照组采用De-Al-HY与异丙醇(★)<sup>[<xref ref-type="bibr" rid="b26">26</xref>]</sup>.(b) SA/70与De-Al-HY催化甘油醛与乙醇反应中催化剂活化前后催化活性的对比.反应条件:1.25 mL含有0.4 mol/L苯甲酰甲醛或甘油醛的醇溶液加入0.05 g催化剂,在363 K下搅拌反应6 h<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Catalytic conversion of C3 sugars over ASAs. (a) Catalytic conversion of phenylglyoxal (―) and selectivity to alkyl mandelate (---) as a function of reaction time in MeOH (■), EtOH (●), <i>i</i>-PrOH (▼), <i>n</i>-PrOH (◆), <i>n</i>-BuOH (▲) over SA/70 and reaction in i-PrOH (★) over De-Al-HY<sup>[<xref ref-type="bibr" rid="b26">26</xref>]</sup>. (b) Catalytic glyceraldehyde conversion in ethanol over fresh De-Al-HY and SA/50 and after five recycle uses. Conditions: 1.25 mL of alcohol solution containing 0.4 mol/L phenylglyoxal or glyceraldehyde, 0.05 g catalyst, at 363 K for 6 h with stirring<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.9
... 在相同条件下,使用二甲苯为溶剂能够产生更高的火焰温度和更快的冷却速度.通过对得到的SA/Xx的27Al MAS NMR谱进行模拟及定量分析,发现以二甲苯为溶剂合成的ASAs能够显著提高AlV的含量[如图 10(a)].与使用甲醇/乙酸(体积比1:1)混合溶剂合成的SA/X相比,使用二甲苯提高AlV含量能够促进AlV与SiOH相互作用,将B酸含量提高了近30%.与传统AlIV富集的ASAs相比,能够将B酸含量提高近10倍[61].特别是使用具有更高燃烧熵的溶剂不仅能够提高AlV含量,还能改善AlV在氧化硅中的分布,促进B酸的形成,因而能在大幅降低铝的用量的条件下合成具有相似酸性的ASAs(如SA/30x和SA/70)[9]. ...
... ,转换频率(TOFs).数据均来自参考文献[9].反应条件见图 9(a) Correlation between the concentration of Brønsted acidic OH groups (BAS, solid line) and Al<sup>V</sup> species (dashed line) as a function of the Al content in dehydrated FSP ASAs prepared by using methanol/acetic acid (1:1 by volume, ■) and xylene (★), respectively; (b) Catalytic conversion of PG in ethanol. The PG conversion (<i>C</i><sub>PG</sub>), EM selectivity (<i>S</i><sub>EM</sub>), reaction rates <i>k</i>, and TOFs were obtained from Ref. [<xref ref-type="bibr" rid="b9">9</xref>], respectively. Same reaction conditions as shown in <xref ref-type="fig" rid="Fig9">Fig. 9</xref>Fig.10
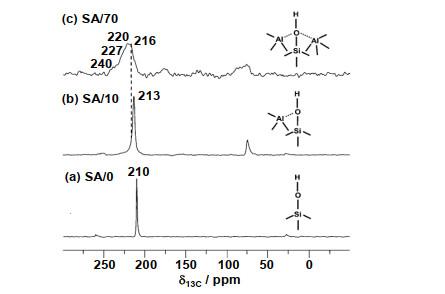
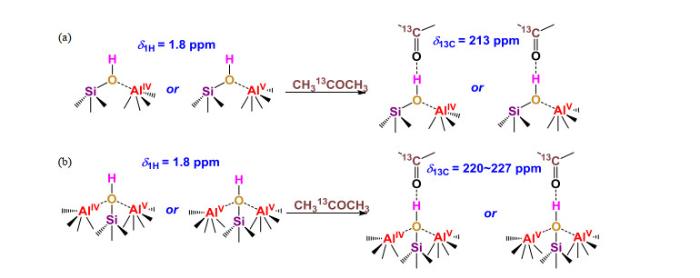
... [10] <sup>13</sup>C MAS NMR spectra of acetone-2-<sup>13</sup>C-loaded Al<sup>V</sup>-enriched ASAs<sup>[<xref ref-type="bibr" rid="b10">10</xref>]</sup>: (a) SA/0; (b) SA/10; (c) SA/70Fig.5(a)~(e) SA/50纳米颗粒的原子探针结构分析;(f)脱水SA/50的二维<sup>27</sup>Al双量子-单量子相关谱(<sup>27</sup>Al DQ-SQ 2D NMR)谱(18.8 T下采集,参数<i>ν</i><sub>R</sub>=20 kHz)及(g)~(k)二维谱中对角峰及交叉峰相应的一维<sup>27</sup>Al NMR谱切片<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>(a)~(e) 3D APT reconstruction of two isolated SA/50 nanoparticles showing all atoms; (f) <sup>27</sup>Al DQ-SQ 2D NMR spectrum recorded at 18.8 T with <i>ν</i><sub>R</sub>=20 kHz of dehydrated SA/50, and (g)~(k) rows extracted from the 2D spectrum corresponding to the various autocorrelation and cross-peaks<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.6
... [10]: (a) SA/0; (b) SA/10; (c) SA/70 Fig.5(a)~(e) SA/50纳米颗粒的原子探针结构分析;(f)脱水SA/50的二维<sup>27</sup>Al双量子-单量子相关谱(<sup>27</sup>Al DQ-SQ 2D NMR)谱(18.8 T下采集,参数<i>ν</i><sub>R</sub>=20 kHz)及(g)~(k)二维谱中对角峰及交叉峰相应的一维<sup>27</sup>Al NMR谱切片<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>(a)~(e) 3D APT reconstruction of two isolated SA/50 nanoparticles showing all atoms; (f) <sup>27</sup>Al DQ-SQ 2D NMR spectrum recorded at 18.8 T with <i>ν</i><sub>R</sub>=20 kHz of dehydrated SA/50, and (g)~(k) rows extracted from the 2D spectrum corresponding to the various autocorrelation and cross-peaks<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.6
... 理论计算认为AlV能够与邻近的SiOH产生相互作用,从而促进表面B酸位的形成[15].但由于缺乏AlV与SiOH相互作用的相关证据,该假设存在广泛争议.首先,常用的一维27Al MAS NMR谱,如1H/27Al双共振实验(TRAPDOR)与27Al{1H}交叉极化(CP)实验,能够提供质子与邻近Al原子的作用信息,但是不能用于区分Al原子的配位[37].二维NMR相关谱广泛用于研究沸石中B酸位的微观结构,同时能够区分不同配位Al原子与邻近质子的相关性[38, 39].但是应用二维NMR相关谱研究ASAs的结构非常困难.这主要是由于ASAs中缺乏长程有序结构、铝的化学环境分布广泛,因而导致探测信号的灵敏度低且相互重叠,特别是难以实现对ASAs中含量极低的AlV的观测. ...
Mechanically induced phase transformation of gamma-Al2O3 into alpha-Al2O3. Access to structurally disordered gamma-Al2O3 with a controllable amount of pentacoordinated Al sites
Highly dispersed SiOx/Al2O3 catalysts illuminate the reactivity of isolated silanol sites
2015
Atomic description of the interface between silica and alumina in aluminosilicates through dynamic nuclear polarization surface-enhanced NMR spectroscopy and first-principles calculations
... [26].(b) SA/70与De-Al-HY催化甘油醛与乙醇反应中催化剂活化前后催化活性的对比.反应条件:1.25 mL含有0.4 mol/L苯甲酰甲醛或甘油醛的醇溶液加入0.05 g催化剂,在363 K下搅拌反应6 h[8]Catalytic conversion of C3 sugars over ASAs. (a) Catalytic conversion of phenylglyoxal (―) and selectivity to alkyl mandelate (---) as a function of reaction time in MeOH (■), EtOH (●), <i>i</i>-PrOH (▼), <i>n</i>-PrOH (◆), <i>n</i>-BuOH (▲) over SA/70 and reaction in i-PrOH (★) over De-Al-HY<sup>[<xref ref-type="bibr" rid="b26">26</xref>]</sup>. (b) Catalytic glyceraldehyde conversion in ethanol over fresh De-Al-HY and SA/50 and after five recycle uses. Conditions: 1.25 mL of alcohol solution containing 0.4 mol/L phenylglyoxal or glyceraldehyde, 0.05 g catalyst, at 363 K for 6 h with stirring<sup>[<xref ref-type="bibr" rid="b8">8</xref>]</sup>Fig.9
... [26]. (b) Catalytic glyceraldehyde conversion in ethanol over fresh De-Al-HY and SA/50 and after five recycle uses. Conditions: 1.25 mL of alcohol solution containing 0.4 mol/L phenylglyoxal or glyceraldehyde, 0.05 g catalyst, at 363 K for 6 h with stirring[8]Fig.9
... Wang等[27]合成了AlV(~48 at.%)富集的ASAs负载的Pt单位点催化剂.相同条件下,Pt主要以团簇及纳米颗粒的形式分布在氧化铝(AlV含量极低)和氧化硅表面.HAADF-STEM及EDS线扫说明单位点Pt的形成依赖于ASAs表面的某种Al位点,而与氧化铝表面的铝位点无明显相关性.如图 11(a)~11(b)所示,负载Pt单位点催化剂后,ASAs表面的AlV含量显著降低,同时有大量的AlIV与AlVI生成.而图 11(c)说明形成Pt团簇及纳米颗粒几乎不会引起AlIV与AlVI的配位变化,这说明单位点Pt的形成仅与AlV的配位变化相关.但是,该27Al MAS NMR谱难以提供合成过程中Pt前驱体(H2PtCl6)与表面AlV作用的信息. ...
... [27] <sup>27</sup>Al MAS NMR spectra of (a) ASA, (b) Pt/ASA, (c) Al<sub>2</sub>O<sub>3</sub> (yellow dash line), and Pt/Al<sub>2</sub>O<sub>3</sub> (blue solid line). Static <sup>27</sup>Al NMR of wet samples obtained after: (d) H<sub>2</sub>PtCl<sub>6</sub> mixed with ASA, (e) mixing (d) with NH<sub>3</sub>•H<sub>2</sub>O, and (f) after heating mixture (e) at 90 ℃ for 1 h, recorded at 11.7 T<sup>[<xref ref-type="bibr" rid="b27">27</xref>]</sup>Fig.11
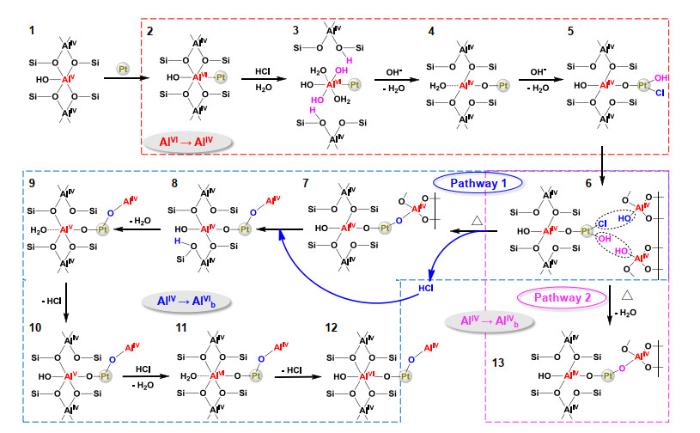
... 依据NMR表征结果,作者提出了AlV诱导Pt单位点催化剂合成的机理(图 13).同时,CO-DRIFTS、XPS及EELS的表征结果揭示了AlV与Pt之间的强相互作用依赖于Al-O-Pt的形成,为基于AlV合成系列单位点催化剂及其性能调控奠定了理论基础.在催化芳族酮选择性加氢反应常用于研究SMSI导致的金属表面电荷变化.研究表明SMSI越强,金属对芳族酮的加氢选择性越差[28, 29, 79].相比于Pt纳米催化剂,尽管ASAs表面的单位点Pt具有更强的SMSI,但是单位点Pt的表面积小,可以有效限制苯环等官能团的吸附及加氢反应.因而,单位点Pt/ASA催化剂展现出显著优于纳米Pt催化剂(Pt/Al2O3、Pt/SiO2)的C=O的加氢选择性(70% vs. 31%~38%).Al<sup>V</sup>诱导Pt单位点催化剂合成的机理<sup>[<xref ref-type="bibr" rid="b27">27</xref>]</sup>Schematic presentation of the proposed pathways for the formation of Pt single sites at Al<sup>V</sup> sites on ASA support<sup>[<xref ref-type="bibr" rid="b27">27</xref>]</sup>Fig.138 总结与展望
... [41],“−60 ppm”和“15 ppm”为17O NMR信号 Optimized structure of Al<sup>V</sup> species in dehydrated states (a, c and e) and in corresponding rehydrated states (b, d, and f), both calculated at B3LYP/6-31g theoretical level<sup>[<xref ref-type="bibr" rid="b6">6</xref>]</sup>; (g) <sup>17</sup>O{<sup>27</sup>Al} TRAPDOR curves obtained from <sup>17</sup>O{<sup>1</sup>H, <sup>27</sup>Al} PRESTO-TRAPDOR experiment were simulated to determine the Al-OH distance, for discriminating the structure of Al<sup>V</sup>-BASs, corresponding to bridging silanols (black line, O-Al distance of 2 Å) and pseudo-bridging silanols (gray shaded area, O-Al distance range of 3 to 4.4 Å)<sup>[<xref ref-type="bibr" rid="b41">41</xref>]</sup>, the corresponding <sup>17</sup>O chemical shifts were at -60 and 15 ppmFig.43 Al<sup>V</sup>富集的ASAs中强B酸位形成的多铝中心协同作用模型
Regioselective H/D exchange at the side-chain of ethylbenzene on dealuminated zeolite H-Y studied by in situ MAS NMR-UV/Vis spectroscopy
4
2008
... 固体酸催化剂中的B酸位能够将质子传递给吸附的碳氢化合物分子,形成碳正离子.碳正离子是碳氢化合物转化的重要反应中间体.通常,强B酸位有助于碳正离子的形成.如沸石中的强酸性桥式羟基能够有效活化苯环上的C-H键,显著提升芳烃的烷基化反应效率[48-51].通过原位1H MAS NMR谱能够研究脱水固体酸表面各种羟基质子与氘代芳烃的相互作用(氢氘交换).通过拟合参与氢氘交换反应的芳烃浓度与时间的关系,可以得到反应的速率曲线.结合不同温度下的速率曲线能够给出活化能等衡量固体酸催化活性的信息. ...
... Wang等[8]通过原位1H MAS NMR谱对比氘代苯与H-ZSM-5沸石及AlV富集的ASAs表面B酸位之间的氢氘反应速率,证明了ASAs中存在强B酸位.与文献报道[48-51]相似,H-ZSM-5沸石表面的酸性羟基(δH 4)含量大幅降低而中性SiOH(δH 1.8)含量基本不变,同时苯环上的质子(δH 7.5)含量显著提高[图 8(a)].这说明了沸石中的酸性桥式羟基是该反应的主要活性位,而中性SiOH不参与反应.但是氢氘反应前后,SA/50表面的SiOH(δH 1.8)含量大幅降低并伴随着苯环上的质子(δH 7.3~7.5)含量显著提高[图 8(b)],这一现象说明SA/50表面的酸性SiOH同样能够催化该反应.通过拟合能够得到了氘代苯与不同固体酸表面B酸位反应的速率k [图 8(c)].一般来说,速率k与B酸位的强度呈正比.该实验中,研究人员通过氘代苯与表面B酸位的1:1吸附及低温扩散后进行测试,更进一步降低了苯的浓度及扩散对实验的影响.该研究表明SA/50与H-ZSM-5沸石具有几乎相同的速率k,远高于SA/10.这说明了SA/50与H-ZSM-5沸石具有相似的B酸强度,DQ-SQ 2D NMR谱以及SA/50的1H MAS NMR谱中没有观测到酸性桥式羟基,SA/50中的强B酸与采用2-13C-丙酮位探针分子的13C CP/MAS NMR谱表征结果[10]一致.结合27Al位被认为是多个不饱和Al原子与同一个SiOH协同作用的结果.这种强B酸位的形成能够显著增强ASAs在一系列C-H键活化反应中的催化活性. ...
In situ H-1 MAS NMR investigations of the H/D exchange of alkylaromatic hydrocarbons on zeolites H-Y, La, Na-Y, and H-ZSM-5
2007
Insight into the mechanisms of the ethylbenzene disproportionation: Transition state shape selectivity on zeolites
2008
Kinetic NMR and density-functional study of benzene H/D exchange in zeolites, The most simple aromatic-substitution
2
1995
... 固体酸催化剂中的B酸位能够将质子传递给吸附的碳氢化合物分子,形成碳正离子.碳正离子是碳氢化合物转化的重要反应中间体.通常,强B酸位有助于碳正离子的形成.如沸石中的强酸性桥式羟基能够有效活化苯环上的C-H键,显著提升芳烃的烷基化反应效率[48-51].通过原位1H MAS NMR谱能够研究脱水固体酸表面各种羟基质子与氘代芳烃的相互作用(氢氘交换).通过拟合参与氢氘交换反应的芳烃浓度与时间的关系,可以得到反应的速率曲线.结合不同温度下的速率曲线能够给出活化能等衡量固体酸催化活性的信息. ...
... Wang等[8]通过原位1H MAS NMR谱对比氘代苯与H-ZSM-5沸石及AlV富集的ASAs表面B酸位之间的氢氘反应速率,证明了ASAs中存在强B酸位.与文献报道[48-51]相似,H-ZSM-5沸石表面的酸性羟基(δH 4)含量大幅降低而中性SiOH(δH 1.8)含量基本不变,同时苯环上的质子(δH 7.5)含量显著提高[图 8(a)].这说明了沸石中的酸性桥式羟基是该反应的主要活性位,而中性SiOH不参与反应.但是氢氘反应前后,SA/50表面的SiOH(δH 1.8)含量大幅降低并伴随着苯环上的质子(δH 7.3~7.5)含量显著提高[图 8(b)],这一现象说明SA/50表面的酸性SiOH同样能够催化该反应.通过拟合能够得到了氘代苯与不同固体酸表面B酸位反应的速率k [图 8(c)].一般来说,速率k与B酸位的强度呈正比.该实验中,研究人员通过氘代苯与表面B酸位的1:1吸附及低温扩散后进行测试,更进一步降低了苯的浓度及扩散对实验的影响.该研究表明SA/50与H-ZSM-5沸石具有几乎相同的速率k,远高于SA/10.这说明了SA/50与H-ZSM-5沸石具有相似的B酸强度,DQ-SQ 2D NMR谱以及SA/50的1H MAS NMR谱中没有观测到酸性桥式羟基,SA/50中的强B酸与采用2-13C-丙酮位探针分子的13C CP/MAS NMR谱表征结果[10]一致.结合27Al位被认为是多个不饱和Al原子与同一个SiOH协同作用的结果.这种强B酸位的形成能够显著增强ASAs在一系列C-H键活化反应中的催化活性. ...
Catalytic arene alkylation over H-Beta zeolite: Influence of zeolite shape selectivity and reactant nucleophilicity
Extra-framework aluminium species in hydrated faujasite zeolite as investigated by two-dimensional solid-state NMR spectroscopy and theoretical calculations
Magic-angle spinning nuclear-magnetic-resonance studies of water-molecules adsorbed on Br?nsted-acid and Lewis-acid sites in zeolites and amorphous silica-aluminas




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}